Chromogenic vs. Fluorescent IHC/ICC: A Complete Guide for Researchers in Biomarker Detection
This article provides a comprehensive comparison of chromogenic (DAB) and fluorescent detection methods in immunohistochemistry (IHC) and immunocytochemistry (ICC).
Chromogenic vs. Fluorescent IHC/ICC: A Complete Guide for Researchers in Biomarker Detection
Abstract
This article provides a comprehensive comparison of chromogenic (DAB) and fluorescent detection methods in immunohistochemistry (IHC) and immunocytochemistry (ICC). It explores the fundamental principles behind each technique, details their specific methodological applications, addresses common troubleshooting and optimization challenges, and offers a critical comparative validation framework. Designed for researchers, scientists, and drug development professionals, this guide synthesizes current best practices to help you select and implement the optimal detection system for your experimental goals, from multiplexing and quantitative analysis to clinical diagnostics.
Core Principles: Understanding the Chemistry and History of Chromogenic and Fluorescent Detection
In the comparative analysis of immunohistochemistry (IHC) and immunocytochemistry (ICC) detection systems, chromogenic detection using 3,3'-Diaminobenzidine (DAB) represents the cornerstone of traditional, bright-field microscopy-based techniques. This whitepaper provides an in-depth technical guide to the core principles and applications of DAB detection, situating its utility within the broader research thesis comparing it to fluorescent methodologies for target visualization in tissues and cells.
Core Principle: The Enzymatic Cascade
Chromogenic DAB detection is an indirect method that utilizes an enzyme, typically Horseradish Peroxidase (HRP), conjugated to a secondary antibody. The HRP catalyzes the oxidation of the DAB chromogen in the presence of hydrogen peroxide (H₂O₂) substrate. This oxidation reaction produces an insoluble, brown-colored precipitate at the site of the target antigen-antibody complex.
Diagram: DAB Chromogenic Signal Generation Pathway
Standard DAB IHC/ICC Protocol: A Detailed Methodology
The following is a generalized step-by-step protocol for DAB detection in formalin-fixed, paraffin-embedded (FFPE) tissue sections, highlighting critical steps for reproducibility.
Protocol:
- Deparaffinization & Rehydration: Incubate slides in xylene (3 changes, 5 min each), followed by a graded ethanol series (100%, 100%, 95%, 70% - 2 min each), and finally distilled water.
- Antigen Retrieval: Perform heat-induced epitope retrieval (HIER) by incubating slides in citrate buffer (pH 6.0) or EDTA buffer (pH 9.0) at 95-100°C for 20 minutes. Cool slides for 30 minutes at room temperature (RT).
- Endogenous Peroxidase Blocking: Incubate slides in 3% H₂O₂ in methanol or PBS for 10 minutes at RT to quench endogenous peroxidase activity. Rinse with wash buffer (e.g., PBS-T).
- Protein Blocking: Apply a non-specific protein block (e.g., 5% normal serum, 1% BSA in PBS) for 30 minutes at RT to reduce background.
- Primary Antibody Incubation: Apply optimally titrated primary antibody diluted in antibody diluent. Incubate in a humidified chamber for 1 hour at RT or overnight at 4°C. Wash 3 x 5 minutes.
- Secondary Antibody Incubation: Apply HRP-conjugated polymer secondary antibody (e.g., from a polymer-based detection kit) for 30 minutes at RT. Wash 3 x 5 minutes.
- Chromogen Application: Prepare DAB substrate solution according to manufacturer's instructions. Apply to tissue sections and monitor development under a microscope (typically 30 seconds to 5 minutes).
- Counterstaining & Mounting: Stop reaction by immersing in distilled water. Counterstain with Hematoxylin for 30-60 seconds. Dehydrate through graded alcohols, clear in xylene, and mount with permanent mounting medium.
Diagram: Standard DAB IHC Experimental Workflow
Quantitative Comparison: Chromogenic DAB vs. Fluorescent Detection
Table 1: Core Characteristics of DAB vs. Fluorescent Detection
| Parameter | Chromogenic (DAB) Detection | Fluorescent Detection |
|---|---|---|
| Detection Mode | Bright-field microscopy | Fluorescence/confocal microscopy |
| Signal Type | Stable, insoluble precipitate | Emitted light (photons) |
| Multiplexing | Limited (sequential, different enzymes/colors) | High (simultaneous, different fluorophores) |
| Sensitivity | High (signal amplification via enzyme) | Very High (amplification possible) |
| Spatial Resolution | Excellent for morphology | Superior for subcellular localization |
| Signal Permanence | Permanent (fade-resistant) | Fades over time (photobleaching) |
| Quantification | Semi-quantitative (density analysis) | Highly quantitative (intensity analysis) |
| Background/ Autofluorescence | Low tissue background | Can be high due to tissue autofluorescence |
| Primary Cost | Lower (standard microscopes) | Higher (fluorescence-capable systems) |
Table 2: Common Research Applications and Suitability
| Research Goal | Preferred Method | Rationale |
|---|---|---|
| Diagnostic Pathology & Morphology | Chromogenic DAB | Permanent stain, excellent contrast with Hematoxylin, standard in clinics. |
| Co-localization Studies (2+ targets) | Fluorescent | Ability to simultaneously visualize multiple targets in the same sample. |
| High-Content, Quantitative Analysis | Fluorescent | Linear signal range enables precise intensity measurement. |
| Archived Tissue Analysis (Long-term) | Chromogenic DAB | Permanent record; no signal degradation over decades. |
| Live or Dynamic Cell Imaging | Fluorescent | Compatible with live-cell imaging and time-lapse studies. |
The Scientist's Toolkit: Essential Reagents for DAB Detection
Table 3: Key Research Reagent Solutions for DAB IHC/ICC
| Reagent / Solution | Function / Purpose | Critical Notes |
|---|---|---|
| Primary Antibody | Binds specifically to the target antigen of interest. | Clone, species, and titer optimization are essential. |
| HRP-Conjugated Polymer Detection System | Links the primary antibody to the HRP enzyme. Provides significant amplification. | Reduces non-specific staining vs. traditional streptavidin-biotin (avoiding endogenous biotin). |
| DAB Chromogen Substrate Kit | Contains DAB and H₂O₂ buffer. The HRP substrate for generating the colored precipitate. | Commercial kits ensure consistency and safety (DAB is a suspected carcinogen). |
| Antigen Retrieval Buffer (Citrate/EDTA) | Reverses formaldehyde-induced cross-links, restoring antibody access to epitopes. | pH and buffer choice are antigen-dependent and require optimization. |
| Blocking Serum (e.g., Normal Goat Serum) | Reduces non-specific binding of secondary antibodies to tissue, lowering background. | Should match the host species of the secondary antibody. |
| Peroxidase Block (3% H₂O₂) | Inhibits endogenous peroxidase activity in tissues (e.g., in red blood cells). | Crucial for preventing false-positive signal. |
| Hematoxylin Counterstain | Provides blue/purple nuclear contrast to the brown DAB signal. | Differentiating (bluing) step is required for optimal nuclear detail. |
| Aqueous or Organic Mounting Medium | Preserves and protects stained tissue for microscopy. | Aqueous for quick viewing; organic (e.g., resin-based) for permanent sealing. |
Chromogenic DAB detection remains a fundamental, robust, and accessible technique, particularly valued for its morphological context, signal permanence, and compatibility with standard pathology workflows. Its strengths lie in single-target analysis, diagnostic applications, and archival studies. Within the broader thesis comparing chromogenic and fluorescent IHC/ICC, DAB is positioned as the gold standard for qualitative to semi-quantitative analysis where spatial relationship to tissue architecture is paramount. However, for advanced multiplexing, precise subcellular localization, and rigorous quantification, fluorescent detection offers distinct advantages. The choice between these core methodologies is therefore contingent on the specific research question, required data output, and available instrumentation.
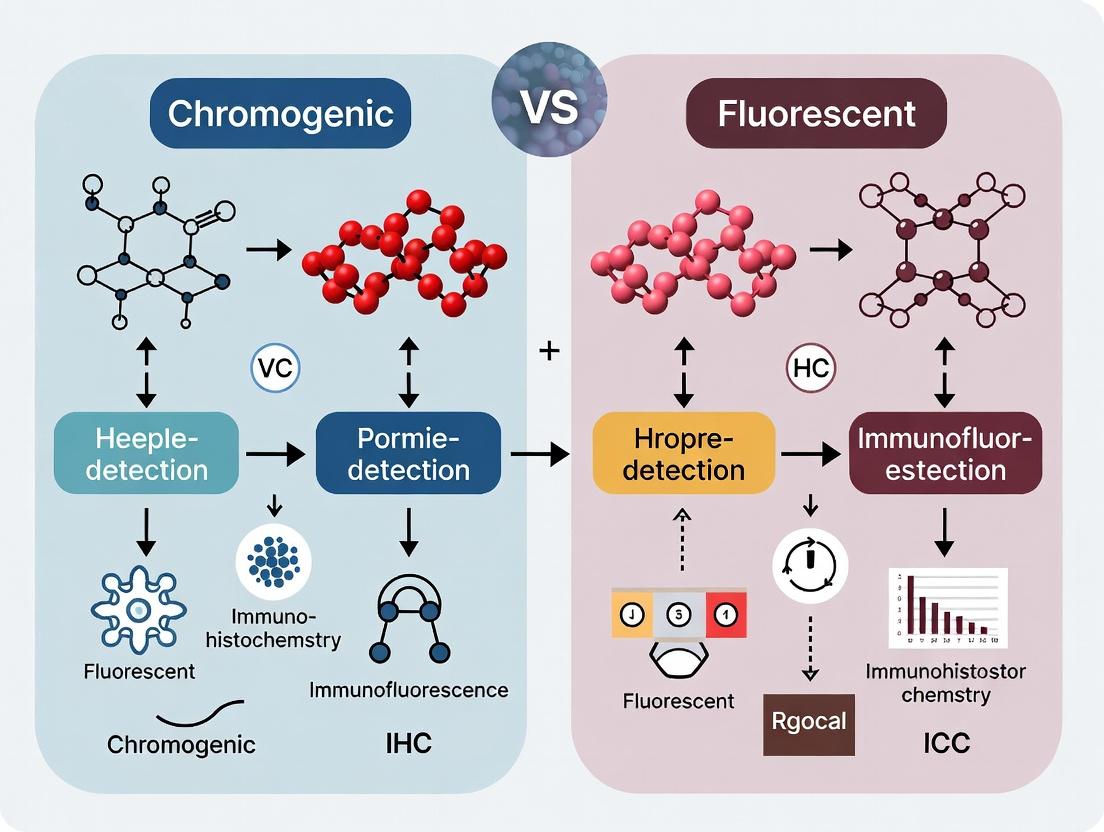
The choice between chromogenic and fluorescent detection methods is foundational to experimental design in immunohistochemistry (IHC) and immunocytochemistry (ICC). This whitepaper details the core principles of fluorescent detection, a cornerstone technique enabling multiplexing, superior sensitivity, and quantitative analysis. The comparative thesis hinges on understanding that while chromogenic detection (colorimetric precipitate) offers permanence and compatibility with brightfield microscopy, fluorescent detection (light emission) provides dynamic range and the capacity for concurrent target analysis, making it indispensable for advanced research and drug development.
Fluorescent detection relies on fluorophores—molecules that absorb high-energy (short-wavelength) photons and subsequently emit lower-energy (longer-wavelength) photons. The core quantitative relationship is described by the Stokes shift, the critical difference between peak excitation and peak emission wavelengths, which allows emitted light to be distinguished from excitation light.
Key Quantitative Parameters of Fluorophores:
| Parameter | Definition | Impact on Experimental Design |
|---|---|---|
| Excitation Maximum (nm) | Wavelength at which absorption is highest. | Determines the required light source/laser line. |
| Emission Maximum (nm) | Wavelength at which light output is highest. | Dictates the choice of emission filter/detector. |
| Stokes Shift (nm) | Difference between emission and excitation maxima. | Larger shifts reduce background (scatter) interference. |
| Extinction Coefficient (M⁻¹cm⁻¹) | Measure of absorption efficiency. | Higher values indicate brighter signal at given concentration. |
| Quantum Yield | Ratio of photons emitted to photons absorbed. | Ranges from 0 to 1; higher yield means brighter fluorophore. |
| Photostability | Resistance to photobleaching upon illumination. | Determines allowable exposure time for image acquisition. |
Signaling Pathways in Fluorescent Detection Systems
Fluorescent detection in IHC/ICC typically employs an antibody conjugated to a fluorophore. For signal amplification, methods like the Tyramide Signal Amplification (TSA) system are used, which leverages horseradish peroxidase (HRP) activity to deposit numerous fluorescent tyramide molecules near the target antigen.
Diagram Title: Tyramide Signal Amplification (TSA) Workflow
Experimental Protocol: Standard Indirect Immunofluorescence for ICC
Objective: To localize a specific protein target within cultured cells using fluorescent detection.
Materials:
- Cells grown on glass coverslips in a multi-well plate.
- Ice-cold 100% methanol or 4% paraformaldehyde (PFA) fixative.
- Phosphate-Buffered Saline (PBS), pH 7.4.
- Permeabilization/Blocking Buffer: PBS with 0.3% Triton X-100 and 5% normal serum from the host species of the secondary antibody.
- Primary antibody specific to the target.
- Fluorophore-conjugated secondary antibody (host species specific to primary antibody).
-
Immunohistochemistry (IHC) and Immunocytochemistry (ICC) are cornerstone techniques for visualizing antigen distribution in tissues and cells. Their evolution is intrinsically linked to the development of detection systems, framed within the enduring research thesis comparing chromogenic versus fluorescent detection. This whitepaper provides a technical guide to this evolution, current state, and practical methodologies.
Historical Timeline & Core Technological Shifts
The progression of detection systems can be categorized into distinct generations, each defined by signal amplification and detection modality.
Table 1: Generations of IHC/ICC Detection Systems
| Generation & Era | Core Technology | Primary Detection Type | Key Advantage | Major Limitation |
|---|---|---|---|---|
| First (1970s) | Direct Conjugation | Fluorescent | Simple, rapid | Low sensitivity, limited multiplexing |
| Second (1980s) | Indirect (Secondary Ab) | Chromogenic/Fluorescent | Signal amplification (~10x), flexibility | Moderate sensitivity, autofluorescence |
| Third (1990s) | Enzyme-Polymer (e.g., HRP-polymer) | Chromogenic | High sensitivity, low background | Signal diffusion, single-plex limitation |
| Fourth (2000s) | Tyramide Signal Amplification (TSA) | Fluorescent/Chromogenic | Extreme sensitivity (>100x), multiplex capable | Optimization complexity, cost |
| Fifth (2010s-Present) | Metal-based Imaging (IMC), Digital AI Analysis | Mass/Fluorescent | High-plex (40+ targets), absolute quantification | Ultra-specialized equipment, data complexity |
Chromogenic vs. Fluorescent Detection: A Quantitative Framework
The core thesis juxtaposes the two primary readout modalities. The choice is not merely aesthetic but dictates experimental design, capabilities, and data output.
Table 2: Chromogenic vs. Fluorescent Detection - A Comparative Analysis
| Parameter | Chromogenic Detection (DAB, AEC) | Fluorescent Detection (Fluorophores, Qdots) |
|---|---|---|
| Signal Type | Precipitation of colored substrate | Emission of light at specific wavelength |
| Readout | Brightfield microscopy | Epifluorescence/Confocal microscopy |
| Multiplexing | Limited (2-3 targets with careful optimization) | High (5+ targets with spectral separation) |
| Sensitivity | Very High (amplified by enzyme kinetics) | High (dependent on fluorophore brightness) |
| Spatial Resolution | Lower (enzyme product diffusion) | High (precise subcellular localization) |
| Permanence | Stable, permanent slides | Prone to photobleaching |
| Tissue Background | Autofluorescence irrelevant; endogenous pigment can interfere | Autofluorescence a major concern |
| Quantification | Semi-quantitative (density based) | Highly quantitative (intensity based) |
| Primary Use Case | Diagnostic pathology, single biomarker | Research, multiplex biomarker discovery, co-localization |
| Common Substrates/Reporters | DAB (brown), AEC (red), HRP/AP enzymes | FITC, TRITC, Alexa Fluor dyes, Quantum Dots |
Detailed Experimental Protocols
Protocol 1: Standard Indirect Chromogenic IHC (DAB)
This is the foundational method for formalin-fixed, paraffin-embedded (FFPE) tissues.
- Deparaffinization & Rehydration: Incubate slides in xylene (2 x 5 min), followed by graded ethanol series (100%, 95%, 70% - 2 min each) to water.
- Antigen Retrieval: Place slides in citrate buffer (pH 6.0) or EDTA (pH 9.0). Heat in pressure cooker (121°C, 15 min) or water bath (95-100°C, 20-40 min). Cool for 30 min.
- Peroxidase Blocking: Incubate with 3% H₂O₂ in methanol for 10 min to quench endogenous peroxidase activity.
- Blocking: Apply 2.5-5% normal serum (from species of secondary antibody) or protein block for 30 min at room temperature (RT).
- Primary Antibody Incubation: Apply optimized dilution of primary antibody in diluent. Incubate at 4°C overnight or 1 hour at RT in a humidified chamber.
- Secondary Antibody Incubation: Apply enzyme-conjugated (HRP) polymer secondary antibody for 30 min at RT.
- Chromogen Development: Apply DAB substrate solution (prepare according to manufacturer's instructions) for 3-10 min. Monitor under microscope. Stop reaction by immersing in distilled water.
- Counterstaining & Mounting: Counterstain with Hematoxylin for 30-60 sec, blue in Scott's tap water. Dehydrate, clear in xylene, and mount with permanent mounting medium.
Protocol 2: Multiplex Fluorescent IHC Using Tyramide Signal Amplification (TSA)
This protocol enables high-sensitivity, sequential multiplexing on a single FFPE section.
- Steps 1-4: As per Protocol 1 (deparaffinization to blocking).
- Primary Antibody Incubation (Round 1): Apply first primary antibody (e.g., Rabbit anti-CD3) overnight at 4°C.
- HRP Polymer Incubation: Apply HRP-conjugated anti-rabbit polymer for 30 min at RT.
- Tyramide-Fluorophore Incubation: Apply Tyramide conjugated to Fluorophore 1 (e.g., Tyramide-Alexa Fluor 488) at 1:100 dilution for 10 min at RT.
- Antigen Stripping/HRP Inactivation: Heat slides in antigen retrieval buffer at 95-100°C for 20 min or use a low pH glycine buffer to denature/dissociate antibodies. Apply 3% H₂O₂ for 10 min to inactivate residual HRP.
- Repeat Cycle: Return to Step 2 of this protocol with the next primary antibody (e.g., Mouse anti-CD8) and a different Tyramide-Fluorophore (e.g., Tyramide-Alexa Fluor 594). Repeat for subsequent markers.
- Counterstaining & Mounting: Counterstain with DAPI (1 µg/mL for 5 min). Rinse and mount with anti-fade mounting medium.
Key Signaling Pathways & Workflows
Chromogenic IHC Signal Generation Pathway
Sequential Multiplex Fluorescent IHC Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Modern IHC/ICC
| Reagent Category | Specific Example(s) | Function & Critical Role |
|---|---|---|
| Detection Kits | HRP/DAB Polymer Kits, Opal TSA Kits | Provides optimized, ready-to-use reagents for specific detection modalities (chromogenic/fluorescent) ensuring sensitivity and reproducibility. |
| Antigen Retrieval Buffers | Citrate (pH 6.0), Tris-EDTA (pH 9.0), EDTA (pH 8.0) | Reverses formaldehyde-induced cross-links to expose epitopes. Buffer pH is antigen-specific and critical for signal intensity. |
| Blocking Reagents | Normal Serum, BSA, Casein, Animal-Free Protein Blocks | Reduces non-specific binding of antibodies to tissue or Fc receptors, lowering background noise. |
| Primary Antibodies | Monoclonal (rabbit, mouse), Recombinant | Specificity is paramount. Validation for IHC/ICC (KO/knockdown confirmed) is essential to avoid false results. |
| Mounting Media | Aqueous Anti-fade (for fluorescence), Permanent Resinous (for DAB) | Preserves signal; anti-fade media retards photobleaching of fluorophores. |
| Automation Platforms | Autostainers (e.g., from Ventana/Leica, Agilent) | Enables standardized, high-throughput, and reproducible staining protocols, vital for clinical and large-scale research. |
| Multispectral Imaging Systems | Vectra Polaris, PhenoImager | Captures full spectral data at each pixel, allowing unmixing of overlapping fluorophores and autofluorescence for true high-plex analysis. |
This technical guide details the core reagents driving chromogenic and fluorescent detection in Immunohistochemistry (IHC) and Immunocytochemistry (ICC). Within the broader thesis comparing chromogenic versus fluorescent detection methodologies, understanding these reagents' properties, performance, and optimal application is paramount for assay design in research and drug development.
Enzymes: The Catalytic Amplifiers
Enzymes conjugate to secondary antibodies or streptavidin to catalyze the conversion of a substrate into a detectable signal. The choice of enzyme is intrinsically linked to the detection mode.
- Horseradish Peroxidase (HRP): The most common enzyme for chromogenic IHC/ICC. It catalyzes the oxidation of a chromogenic substrate in the presence of hydrogen peroxide (H₂O₂), yielding an insoluble, colored precipitate at the antigen site. Sensitive to endogenous peroxidase activity, requiring inhibition steps.
- Alkaline Phosphatase (AP): An alternative to HRP, often used in multiplexing or when endogenous peroxidase activity is high. It catalyzes the removal of phosphate groups from substrates, leading to colored or fluorescent products. Sensitive to endogenous phosphatase activity.
- Key Considerations: Enzyme size impacts penetration (HRP is smaller than AP). Activity is influenced by pH, inhibitors, and the choice of substrate-buffer system.
Chromogens: The Color Generators
Chromogens are enzyme substrates that yield a visible, localized precipitate upon enzymatic conversion.
Table 1: Common Chromogens in IHC/ICC
| Chromogen (Enzyme) | Final Color | Solubility | Compatible Counterstain | Notes |
|---|---|---|---|---|
| 3,3'-Diaminobenzidine (DAB) (HRP) | Brown | Insoluble | Hematoxylin | Gold standard; permanent; can be enhanced with metals (e.g., nickel, cobalt). |
| 3-Amino-9-ethylcarbazole (AEC) (HRP) | Red | Alcohol-soluble | Hematoxylin, Methyl Green | Requires aqueous mounting; fades over time. |
| Vector VIP (HRP) | Purple | Insoluble | None or light counterstain | High contrast; good for low-abundance antigens. |
| Vector NovaRED (HRP) | Reddish-brown | Insoluble | Hematoxylin | Alternative to AEC with better permanence. |
| 5-Bromo-4-chloro-3-indolyl phosphate/Nitro blue tetrazolium (BCIP/NBT) (AP) | Blue/Black | Insoluble | Nuclear Fast Red, Eosin | Avoid with endogenous AP; good for multiplex. |
| Fast Red (AP) | Red | Alcohol-soluble | Hematoxylin | Fluorescent under certain conditions; aqueous mounting. |
Fluorophores: The Light Emitters
Fluorophores are molecules that absorb light at a specific wavelength and emit light at a longer wavelength. They are conjugated directly to primary antibodies (direct fluorescence) or, more commonly, to secondary antibodies/streptavidin (indirect fluorescence).
Table 2: Common Fluorophores in IHC/ICC
| Fluorophore | Excitation (nm) Max | Emission (nm) Max | Relative Brightness* | Photostability | Common Applications |
|---|---|---|---|---|---|
| DAPI (Counterstain) | 358 | 461 | N/A | Moderate | Nuclear counterstain. |
| FITC | 495 | 519 | 1.0 (Reference) | Low | Common green channel; prone to fading. |
| Alexa Fluor 488 | 495 | 519 | ~1.5-2.0x FITC | High | Superior green alternative to FITC. |
| TRITC | 557 | 576 | ~0.5x FITC | Moderate | Traditional red-orange. |
| Alexa Fluor 555 | 555 | 565 | ~2.0x TRITC | High | Bright red-orange; common for 561nm laser. |
| Cy3 | 550 | 570 | ~1.5x TRITC | Moderate | Common alternative to TRITC. |
| Texas Red | 595 | 615 | ~0.8x FITC | Moderate | Red emission; good for multiplex. |
| Alexa Fluor 647 | 650 | 665 | High | Very High | Far-red; low autofluorescence in tissue. |
| Cy5 | 649 | 670 | High | High | Common far-red channel. |
*Brightness is a product of extinction coefficient and quantum yield, compared roughly to FITC.
Counterstains: Providing Context
Counterstains provide histological context by staining cellular or tissue structures not targeted by the primary antibody.
Table 3: Common Counterstains for Chromogenic and Fluorescent Detection
| Counterstain | Detection Mode | Target | Color | Function & Notes |
|---|---|---|---|---|
| Hematoxylin | Chromogenic | DNA (Chromatin), RNA | Blue | Nuclear stain; different "bluing" agents (ammonia water, Scott's tap water) affect hue. |
| Nuclear Fast Red | Chromogenic | DNA, Calcium | Red/Pink | Nuclear or cytoplasmic; alternative to hematoxylin for red chromogens. |
| Methyl Green | Chromogenic | DNA | Green | Specific for double-stranded DNA; used with red chromogens. |
| DAPI | Fluorescent | DNA (AT-rich regions) | Blue | Gold standard nuclear counterstain for fluorescence; UV excitation. |
| Hoechst 33342 | Fluorescent | DNA (Minor groove) | Blue | Live-cell compatible; permeant. Common for ICC. |
| Propidium Iodide (PI) | Fluorescent | DNA/RNA | Red | Impermeant; stains dead cells or requires permeabilization. |
| Eosin | Chromogenic | Proteins (Cytoplasm) | Pink | Cytoplasmic stain; provides tissue morphology in H&E. |
| SYTO RNASelect | Fluorescent | RNA | Green | Specific for cytoplasmic RNA; useful for cellular delineation. |
Experimental Protocols
Standard Chromogenic IHC Protocol (Indirect, HRP-DAB)
This protocol is for formalin-fixed, paraffin-embedded (FFPE) tissue sections.
- Deparaffinization & Rehydration: Xylene (2 x 5 min) → 100% Ethanol (2 x 2 min) → 95% Ethanol (2 min) → 70% Ethanol (2 min) → dH₂O (2 min).
- Antigen Retrieval: Place slide in pre-heated citrate buffer (pH 6.0) or EDTA/Tris-EDTA buffer (pH 9.0). Heat in pressure cooker, steamer, or water bath (95-100°C) for 10-20 min. Cool for 30 min at room temperature (RT). Rinse in dH₂O.
- Endogenous Peroxidase Blocking: Incubate with 3% H₂O₂ in methanol or PBS for 10 min at RT. Rinse with wash buffer (PBS + 0.025% Triton X-100).
- Blocking: Apply 2-5% normal serum (from species of secondary antibody) or protein block for 30 min at RT.
- Primary Antibody Incubation: Apply optimally diluted primary antibody in diluent. Incubate in a humidified chamber for 1 hr at RT or overnight at 4°C. Wash (3 x 5 min).
- Secondary Antibody Incubation: Apply HRP-conjugated secondary antibody (e.g., anti-rabbit HRP) for 30 min at RT. Wash (3 x 5 min).
- Signal Development: Prepare DAB substrate according to manufacturer's instructions. Apply to tissue and monitor development under a microscope (typically 30 sec - 5 min). Stop reaction by immersing in dH₂O.
- Counterstaining: Immerse in Hematoxylin for 30-60 sec. Rinse in tap water. Differentiate in 1% acid alcohol (1 sec dip) if needed. "Blue" in Scott's tap water or ammonia water. Rinse.
- Dehydration & Mounting: 70% EtOH (30 sec) → 95% EtOH (30 sec) → 100% EtOH (2 x 30 sec) → Xylene (2 x 2 min). Mount with permanent mounting medium (e.g., DPX).
Standard Fluorescent ICC Protocol (Indirect)
This protocol is for cultured cells fixed on coverslips.
- Fixation: Aspirate media. Rinse with PBS. Fix with 4% paraformaldehyde in PBS for 10 min at RT. Rinse (3 x 5 min) with PBS.
- Permeabilization & Blocking: Incubate with blocking buffer (PBS containing 3-5% BSA and 0.3% Triton X-100) for 60 min at RT.
- Primary Antibody Incubation: Apply primary antibody diluted in antibody dilution buffer (PBS with 1% BSA, 0.1% Triton X-100) overnight at 4°C in a humidified chamber. Wash (3 x 5 min) with PBS-T (PBS + 0.1% Tween-20).
- Secondary Antibody Incubation: Apply fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 555 anti-mouse) diluted in antibody dilution buffer. Incubate for 60 min at RT in the dark. Wash (3 x 5 min) with PBS-T in the dark.
- Counterstaining & Mounting: Incubate with DAPI (300 nM in PBS) for 5 min at RT. Wash with PBS (2 x 5 min). Blot excess liquid and mount coverslip onto slide using antifade mounting medium (e.g., ProLong Diamond). Seal with nail polish. Store slides at 4°C in the dark.
Diagrammatic Representations
Diagram 1: Core Detection Pathways for IHC and ICC
Diagram 2: Decision and Workflow for IHC/ICC Detection Methods
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for IHC/ICC Experiments
| Category | Reagent/Material | Function | Key Considerations |
|---|---|---|---|
| Sample Prep | Formalin, Paraformaldehyde (PFA) | Cross-linking fixative for preserving tissue/cell morphology and antigenicity. | Fixation time and concentration are critical; over-fixation masks antigens. |
| Paraffin, OCT Compound | Embedding media for FFPE or frozen tissue sectioning, respectively. | OCT is for cryosectioning; ensures structural integrity during cutting. | |
| Retrieval/Buffers | Citrate Buffer (pH 6.0), Tris-EDTA (pH 9.0) | Antigen retrieval solutions to reverse formaldehyde cross-linking (epitope unmasking). | pH choice depends on the target antigen; heat method (pressure cooker, steamer) must be consistent. |
| Phosphate-Buffered Saline (PBS) | Isotonic washing and dilution buffer. Maintains pH and osmolarity. | Add detergents (Tween-20, Triton X-100) for washing and permeabilization. | |
| Blocking/Detection | Normal Serum (e.g., Goat, Donkey) | Blocks non-specific binding of secondary antibodies to tissue/cells. | Should match the host species of the secondary antibody. |
| Bovine Serum Albumin (BSA) | General blocking agent to reduce non-specific background. | Used at 1-5% in buffers for blocking and antibody dilution. | |
| Antibodies | Validated Primary Antibodies | Specific recognition of the target antigen. | Validation for IHC/ICC specific application is essential. |
| Enzyme- or Fluorophore-conjugated Secondary Antibodies | Amplifies signal and provides detection modality. | Must be raised against the host species of the primary antibody. | |
| Signal Generation | DAB, AEC, BCIP/NBT Kits | Chromogenic substrates for HRP or AP enzymes. | Kits provide stable, optimized substrate-buffer mixtures. |
| Alexa Fluor, Cy Dye Conjugates | Synthetic fluorophores for fluorescent detection. | Superior brightness and photostability compared to traditional dyes (FITC, TRITC). | |
| Counterstains/Mounting | Hematoxylin, DAPI, Hoechst | Provides histological/cytological context by staining nuclei. | DAPI/Hoechst for fluorescence; Hematoxylin for brightfield. |
| Antifade Mounting Medium (e.g., ProLong) | Preserves fluorescence and retards photobleaching. | Critical for long-term storage of fluorescent samples. | |
| Coverslips, Slide Sealant (Nail Polish) | Protects specimen and provides correct optical path for microscopy. | Use #1.5 thickness coverslips for high-resolution oil immersion objectives. |
The Critical Role of Detection Sensitivity and Signal-to-Noise Ratio
Within the comparative research of chromogenic (DAB) versus fluorescent detection in Immunohistochemistry (IHC) and Immunocytochemistry (ICC), the core technical determinants of assay success are detection sensitivity and signal-to-noise ratio (SNR). Sensitivity defines the lowest concentration of a target antigen that can be reliably detected. SNR quantifies the magnitude of the specific signal relative to background, non-specific staining. The choice between chromogenic and fluorescent detection directly impacts these parameters, influencing the accuracy, reproducibility, and quantitative potential of experimental data in research and drug development.
Fundamental Principles: Sensitivity and SNR
Detection Sensitivity is a function of the amplification capability of the detection system and the label's detectability. Signal-to-Noise Ratio is the critical measure of assay fidelity, calculated as (Signal Intensity of Target - Background Intensity) / Standard Deviation of Background.
Fluorescent systems typically offer higher potential sensitivity due to the absence of a quenching step and the ability to detect single photons. However, autofluorescence from tissues or cells can severely degrade SNR. Chromogenic detection, through enzyme-mediated precipitation, provides excellent spatial resolution and permanence but may have lower dynamic range and can be limited by enzyme kinetics and endogenous enzyme activity.
Quantitative Comparison of Chromogenic vs. Fluorescent Detection
The following table summarizes key performance metrics based on recent literature and product data sheets.
Table 1: Comparative Analysis of Chromogenic and Fluorescent Detection for IHC/ICC
| Parameter | Chromogenic Detection (e.g., HRP-DAB) | Fluorescent Detection (e.g., Direct/Indirect IF) | Impact on Sensitivity/SNR |
|---|---|---|---|
| Detection Limit | ~100-1000 copies/cell (dependent on amplification) | ~10-100 copies/cell (with high-quality reagents) | Fluorescence is inherently more sensitive for low-abundance targets. |
| Signal Amplification | High (via enzyme-catalyzed precipitation) | Variable (direct: low; indirect: medium; tyramide: high) | Enzymatic chromogen precipitation offers robust, user-friendly amplification. |
| Background Sources | Endogenous peroxidase, nonspecific antibody binding, incomplete blocking. | Autofluorescence, nonspecific antibody binding, spectral bleed-through. | Autofluorescence is a major SNR challenge for fluorescence, especially in formalin-fixed tissue. |
| Dynamic Range | Narrow (signal saturates, nonlinear) | Wide (linear over several orders of magnitude) | Fluorescence is superior for quantification and co-localization studies. |
| Multiplexing Capacity | Low (sequential staining, color separation challenging) | High (simultaneous detection of multiple targets) | Fluorescence enables complex pathway analysis within a single sample. |
| Quantitation | Semi-quantitative via image densitometry | Highly quantitative via fluorescence intensity | Direct link between SNR and quantitative accuracy favors fluorescence. |
Experimental Protocols for SNR Optimization
Protocol: Minimizing Background in Fluorescent IHC (Indirect Method)
Objective: To achieve high SNR for a low-abundance membrane protein in formalin-fixed, paraffin-embedded (FFPE) tissue.
- Deparaffinization & Antigen Retrieval: Slide baking (60°C, 1 hr), xylene, graded ethanol. Perform heat-induced epitope retrieval (HIER) in citrate buffer (pH 6.0) at 95-100°C for 20 min.
- Autofluorescence Quenching: Treat sections with 0.1% Sudan Black B in 70% ethanol for 15 min. Rinse thoroughly.
- Blocking: Incubate with protein block (5% normal serum, 1% BSA, 0.1% Triton X-100 in PBS) for 1 hr.
- Primary Antibody: Incubate with target-specific monoclonal antibody (optimized dilution in antibody diluent) at 4°C overnight.
- Secondary Antibody: Apply fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 594, 1:500) for 1 hr at RT in the dark.
- Counterstain & Mount: Apply DAPI (1 µg/mL, 5 min), mount with autofluorescence-reducing mounting medium.
- Imaging: Acquire images using a fluorescence microscope with appropriate filter sets. Capture control slides (no primary antibody) to establish background intensity.
Protocol: Enhancing Sensitivity in Chromogenic IHC (Polymer-Based HRP)
Objective: To detect a nuclear transcription factor with high sensitivity in FFPE tissue.
- Deparaffinization & Antigen Retrieval: As per 4.1.
- Endogenous Peroxidase Block: Incubate with 3% H₂O₂ in methanol for 15 min.
- Blocking: Incubate with serum block (10% normal goat serum) for 30 min.
- Primary Antibody: Apply rabbit polyclonal antibody (optimized dilution) for 1 hr at RT.
- Polymer Detection: Apply anti-rabbit HRP-labeled polymer for 30 min.
- Chromogen Development: Incubate with DAB+ chromogen/substrate solution. Monitor development under a microscope (2-10 min). Stop reaction in dH₂O.
- Counterstain & Mount: Hematoxylin counterstain, dehydrate, clear, and mount with permanent mounting medium.
- Image Analysis: Use brightfield microscopy. Quantitative analysis can be performed using positive pixel count algorithms on scanned slides.
Visualizing Key Concepts and Workflows
Figure 1: Determinants of Signal-to-Noise Ratio in IHC/ICC
Figure 2: Comparative Workflow for Detection Methods
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Optimizing Sensitivity and SNR
| Reagent / Solution | Primary Function | Application Note |
|---|---|---|
| High-Specificity, Validated Primary Antibodies | Binds target antigen with minimal off-target interaction. | The single greatest factor in determining SNR. Use antibodies validated for IHC/ICC in your species/tissue. |
| Polymer-Based Detection Systems (HRP/AP) | Provides high-amplification, enzyme-linked secondary detection. | Reduces nonspecific binding vs. traditional avidin-biotin. Crucial for low-abundance targets in chromogenic IHC. |
| Tyramide Signal Amplification (TSA) Kits | Enzyme-driven deposition of fluorophore or hapten tyramides for extreme signal amplification. | Dramatically boosts sensitivity for both fluorescence and chromogenic detection, ideal for challenging targets. |
| Autofluorescence Quenchers (e.g., Sudan Black, TrueBlack) | Reduces background fluorescence from fixatives (like glutaraldehyde) and endogenous biomolecules. | Essential pre-treatment for fluorescent IHC/ICC on FFPE tissues to improve SNR. |
| Antigen Retrieval Buffers (Citrate, EDTA, Tris-EDTA) | Reverses formaldehyde-induced cross-links to expose epitopes. | Critical for FFPE samples. pH and buffer choice must be optimized for each target antigen. |
| Fluorophore-Conjugated Secondaries (e.g., Alexa Fluor, DyLight) | Highly stable, bright fluorophores for direct detection of primaries. | Choice of fluorophore affects sensitivity and multiplexing potential due to extinction coefficient and quantum yield. |
| Phenolic Compound-Free Mounting Media (Fluorescence) | Preserves fluorophore signal, reduces photobleaching, often contains DAPI. | Use hard-set for permanence or aqueous for short-term. Essential for preserving high SNR during imaging/storage. |
| Chromogen Substrates (DAB, AEC, Vector NovaRED) | Enzyme substrate that yields an insoluble colored precipitate at the antigen site. | DAB is most common (brown, permanent). Choice affects contrast and compatibility with counterstains. |
Choosing Your Method: When and How to Apply Chromogenic or Fluorescent IHC/ICC
Within the broader research thesis comparing chromogenic (IHC) and fluorescent (ICC) detection methodologies, chromogenic immunohistochemistry remains the undisputed cornerstone for formalin-fixed, paraffin-embedded (FFPE) tissue analysis in specific, high-value applications. Its compatibility with brightfield microscopy, permanent staining, and straightforward integration into established histopathology workflows cement its role in clinical diagnostics and morphological research. This technical guide details the ideal use cases, protocols, and quantitative data underpinning chromogenic IHC's enduring utility.
Core Applications and Quantitative Advantages
Chromogenic IHC is the method of choice in applications where co-localization with traditional histology, high throughput, and cost-effectiveness are paramount.
Table 1: Quantitative Comparison of IHC Detection Modalities in Key Applications
| Application Domain | Preferred Method | Key Quantitative Metric | Typical Chromogenic Performance | Rationale for Preference |
|---|---|---|---|---|
| Clinical Diagnostic Pathology | Chromogenic IHC | Signal Stability (Archival) | >10 years | Permanent stain for patient records; compatible with H&E counterstain. |
| Surgical Margin Assessment (Intraoperative) | Chromogenic IHC | Assay Time-to-Result | 20-30 minutes (Fast kits) | Rapid, interpretable on standard brightfield scopes in OR. |
| High-Throughput Biomarker Screening (FFPE TMAs) | Chromogenic IHC | Slides Processed per Day | 100-500+ (with automation) | Lower cost per slide; no quenching; easy batch scanning. |
| Multiplexing (2-4 markers) | Chromogenic IHC (Sequential) | Successful Co-localization Rate | >95% (with optimized stripping) | Clear spatial context on single slide; sequential visualization avoids spectral overlap. |
| High-plex Multiplexing (5+ markers) | Fluorescent ICC | Channels Resolved Simultaneously | Limited to ~4 | Chromogenic color separation becomes limiting; fluorescence offers superior multiplexity. |
Detailed Experimental Protocol: Sequential Multiplex Chromogenic IHC
This protocol is critical for evaluating co-expression and spatial relationships of 2-3 biomarkers within the same tissue section, a common need in oncology research.
Materials:
- FFPE tissue section (4-5 µm) on charged slide
- Target Retrieval Buffer (Citrate, pH 6.0 or EDTA/TRIS, pH 9.0)
- Primary Antibodies: Mouse monoclonal [Target A], Rabbit monoclonal [Target B]
- HRP-conjugated Secondary Antibodies (e.g., anti-mouse and anti-rabbit)
- Chromogen Substrates: DAB (brown), Vector Red (magenta), Vector Blue (blue).
- Antibody Elution Buffer (e.g., glycine-HCl, pH 2.0, or commercial stripping buffer)
- Hematoxylin counterstain
Procedure:
- Deparaffinization & Antigen Retrieval: Bake slide, deparaffinize in xylene, rehydrate through graded alcohols. Perform heat-induced epitope retrieval in appropriate buffer using a pressure cooker or steamer for 15-20 min. Cool and rinse.
- Endogenous Peroxidase Block: Incubate with 3% H₂O₂ for 10 min. Wash.
- Protein Block: Apply serum or protein block for 10 min.
- First Immunoreaction:
- Apply primary antibody for [Target A]. Incubate (60 min, RT or overnight, 4°C).
- Wash. Apply HRP-conjugated anti-mouse polymer. Incubate 30 min.
- Wash. Apply DAB chromogen. Develop for 3-10 min (monitor microscopically).
- Rinse in distilled water.
- Antibody Elution:
- Immerse slide in pre-warmed (95°C) antibody elution buffer for 20-30 min.
- Cool and wash thoroughly. Validation of complete elution is essential by omitting the second primary antibody in a control slide.
- Second Immunoreaction:
- Repeat steps 3-4 for [Target B] using a different chromogen (e.g., Vector Red).
- Use a secondary polymer system with different species specificity to avoid cross-reactivity.
- Counterstaining & Mounting: Apply hematoxylin for 30-60 sec. Rinse, blue in Scott's tap water. Dehydrate, clear, and mount with non-aqueous mounting medium.
Sequential Multiplex Chromogenic IHC Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Robust Chromogenic IHC
| Item | Function & Critical Consideration |
|---|---|
| Polymer-based HRP Detection System | Amplifies signal with high sensitivity and low background. Superior to streptavidin-biotin (Avidin-Biotin Complex - ABC) due to lack of endogenous biotin interference. |
| DAB (3,3'-Diaminobenzidine) Chromogen | Forms an insoluble, stable brown precipitate. The gold standard; requires careful hazard management as a potential carcinogen. |
| Alternative Chromogens (Red, Blue) | Enable multiplexing. Must be soluble in organic solvents for dehydration and stable under coverslipping. |
| Antigen Retrieval Buffers | Reverses formaldehyde-induced cross-links. pH and buffer chemistry (citrate vs. EDTA/TRIS) must be optimized for each target epitope. |
| Antibody Elution Buffer | For sequential multiplexing. Must remove primary/secondary antibodies without damaging tissue morphology or remaining antigens. |
| Automated Staining Platform | Provides unparalleled reproducibility and throughput for clinical and large-scale research studies. Standardizes incubation times and temperatures. |
Signaling Pathway Visualization in Key Diagnostic Markers
The interpretation of chromogenic IHC hinges on understanding the pathway context of the detected biomarker.
Key Signaling Pathways Targeted in Diagnostic IHC
In the comparative landscape of detection techniques, chromogenic IHC is not a legacy technology but a specialized one. Its ideal applications—routine clinical diagnostics, surgical pathology, histopathology research, and low-plex multiplexing—leverage its strengths of permanence, morphological context, and seamless integration into the brightfield microscopy ecosystem. While fluorescent ICC is indispensable for high-plex spatial biology and quantification, chromogenic IHC provides the accessible, robust, and legally-definitive foundation for tissue-based diagnosis and biomarker validation.
Immunohistochemistry (IHC) and immunocytochemistry (ICC) are cornerstone techniques for visualizing target protein expression and localization within tissues and cells. The broader thesis comparing chromogenic (colorimetric) and fluorescent detection methodologies reveals fundamental trade-offs. Chromogenic detection, typically using enzymes like horseradish peroxidase (HRP) with 3,3'-Diaminobenzidine (DAB), offers permanent slides compatible with brightfield microscopy but is inherently limited to 1-2 targets due to color crosstalk. Fluorescent detection, using fluorophore-conjugated antibodies, enables multiplexing—the simultaneous detection of multiple targets on a single sample. This guide details the technical execution of fluorescent multiplex IHC/ICC, a critical methodology for understanding complex cellular interactions, cell phenotypes, and spatial biology in research and drug development.
Principles of Fluorescent Multiplexing
Multiplexing relies on the use of non-overlapping fluorescent labels. Key principles include:
- Spectral Separation: Each fluorophore must have distinct excitation and emission spectra. The number of simultaneous targets is limited by the available spectral bandwidth of the detection system (filter sets or spectral imagers).
- Antibody Validation: Primary antibodies must be raised in different host species or be of different isotypes to prevent cross-reactivity from secondary detection. Alternatively, direct conjugation of fluorophores to primary antibodies (direct fluorescence) eliminates secondary antibody concerns.
- Signal Amplification: While direct fluorescence is simple, tyramide signal amplification (TSA) systems can dramatically increase sensitivity for low-abundance targets.
Quantitative Comparison: Chromogenic vs. Fluorescent IHC/ICC
Table 1: Core Comparison of Chromogenic and Fluorescent Detection Methods
| Feature | Chromogenic IHC/ICC | Fluorescent IHC/ICC (Multiplex) |
|---|---|---|
| Max Targets/Slide (Routine) | 1-2 | 4-8+ (with standard filter sets); 30+ with spectral unmixing |
| Detection Method | Enzyme (HRP/AP) → precipitating chromogen | Fluorophore emission |
| Microscope | Brightfield | Epifluorescence, Confocal, Multiphoton, Spectral |
| Permanence of Signal | High (stain is permanent) | Low-Medium (fluorophores can bleach) |
| Compatibility with H&E | Excellent (sequential staining common) | Poor (fluorescence obscured by hematoxylin) |
| Quantitative Analysis | Semi-quantitative (density based) | Highly Quantitative (intensity based) |
| Spatial Resolution | Cellular/Subcellular | Subcellular (confocal) |
| Primary Antibody Host Species | Critical for multiplexing | Critical for multiplexing |
| Key Advantage | Permanent record, pathologist familiarity | Multiplexing, co-localization, quantitative depth |
| Key Limitation | Limited multiplexing, color crosstalk | Autofluorescence, photobleaching, complex analysis |
Table 2: Common Fluorophores for Multiplexing (Exemplary Set)
| Fluorophore | Excitation (nm) Max | Emission (nm) Max | Common Laser Lines (nm) | Recommended For |
|---|---|---|---|---|
| DAPI (Nuclear stain) | 358 | 461 | 405 | Counterstain |
| Alexa Fluor 488 | 495 | 519 | 488 | High-intensity, 1st target |
| Cy3 / Alexa Fluor 555 | 550 | 570 | 543, 561 | 2nd target |
| Alexa Fluor 594 | 590 | 617 | 594 | 3rd target |
| Alexa Fluor 647 | 650 | 665 | 633, 647 | 4th target (low autofluorescence) |
| Cy5 | 649 | 670 | 633, 647 | Deep-red channel |
Detailed Experimental Protocols
Protocol 1: Standard Multiplex Fluorescent IHC (Indirect, 4-Color)
This protocol uses secondary antibodies for signal generation and amplification.
Materials: See "The Scientist's Toolkit" below. Sample: Formalin-fixed, paraffin-embedded (FFPE) tissue sections (4-5 µm) on charged slides.
Procedure:
- Deparaffinization & Antigen Retrieval:
- Bake slides at 60°C for 20 min.
- Deparaffinize in xylene (2 x 5 min) and rehydrate through graded ethanol (100%, 95%, 70%) to distilled water.
- Perform heat-induced epitope retrieval (HIER) in citrate buffer (pH 6.0) or Tris-EDTA buffer (pH 9.0) using a pressure cooker or steamer for 15-20 min.
- Cool slides for 30 min at room temperature (RT), wash in PBS (pH 7.4).
Peroxidase Blocking & Permeabilization (if needed):
- Incubate with 3% H₂O₂ in PBS for 10 min to quench endogenous peroxidase (critical if using TSA).
- Wash in PBS (2 x 5 min).
- For ICC or membrane targets, permeabilize with 0.1% Triton X-100 in PBS for 10 min. Wash.
Protein Blocking:
- Incubate with 5-10% normal serum (from the species of your secondary antibodies) or a commercial protein block for 30-60 min at RT to reduce non-specific binding.
Primary Antibody Incubation (Sequential or Mixed Cocktail):
- Option A (Sequential): Apply first primary antibody (e.g., mouse anti-CD8) diluted in antibody diluent. Incubate overnight at 4°C in a humidified chamber.
- Wash in PBS + 0.025% Tween 20 (PBST) (3 x 5 min).
- Apply species-specific HRP-conjugated secondary antibody (e.g., anti-mouse-HRP) for 60 min at RT. Wash.
- Apply fluorophore-conjugated tyramide (e.g., Alexa Fluor 488-Tyramide) for 10 min. Wash.
- Antibody Stripping (Optional but recommended for high-abundance targets): Perform heat or chemical stripping (e.g., 20min in glycine-HCl buffer, pH 2.0) to remove the primary-secondary complex before next round. Wash thoroughly.
- Repeat cycle for second target (e.g., rabbit anti-CD68 → anti-rabbit-HRP → Cy3-Tyramide).
- Option B (Mixed Cocktail): If validated and host species don't cross-react, apply a cocktail of directly fluorophore-conjugated primary antibodies simultaneously overnight.
Nuclear Counterstain & Mounting:
- Incubate with DAPI (300 nM in PBS) for 5 min.
- Wash in PBS (2 x 5 min).
- Coverslip using a commercial anti-fade mounting medium (e.g., ProLong Diamond).
Imaging:
- Image using an epifluorescence microscope with appropriate filter sets or a confocal microscope. Acquire images sequentially (channel by channel) to minimize bleed-through.
Protocol 2: Multiplex ICC for Cultured Cells (Direct Conjugation)
A simpler protocol ideal for co-localization studies in cells.
Sample: Cells grown on chambered coverslips, fixed (4% PFA, 15 min) and permeabilized (0.1% Triton X-100, 10 min).
Procedure:
- Block with 5% BSA in PBS for 30 min.
- Prepare a cocktail of directly fluorophore-conjugated primary antibodies (e.g., anti-α-Tubulin-AF488, anti-Mitochondria-AF594, anti-Phalloidin-AF647 for F-actin) in blocking buffer.
- Apply the cocktail to cells. Incubate for 2 hours at RT or overnight at 4°C in the dark.
- Wash with PBST (3 x 5 min).
- Counterstain nuclei with DAPI and mount.
Visualization: Signaling Pathways and Workflows
Diagram 1: Sequential TSA Multiplex IHC Workflow
Diagram 2: Core Functional Comparison of IHC Methods
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Fluorescent Multiplex IHC/ICC
| Item | Function & Rationale | Example Product Types |
|---|---|---|
| Validated Primary Antibodies | High specificity for target antigen. Critical for multiplexing: must be from different host species or directly conjugated. | Rabbit monoclonal, Mouse monoclonal, Guinea pig polyclonal. |
| Fluorophore-Conjugated Secondaries or TSA Kits | Generate fluorescent signal. TSA kits provide high sensitivity for low-abundance targets. | Alexa Fluor-conjugated secondaries, Opal TSA kits, Tyramide SuperBoost kits. |
| Antibody Diluent | Optimized buffer to maintain antibody stability and reduce background. | Commercial antibody diluents with carrier proteins and stabilizers. |
| Autofluorescence Quenchers | Reduce tissue/cell autofluorescence, especially in green channel, improving signal-to-noise. | Vector TrueVIEW, Sudan Black B, copper sulfate. |
| Anti-Fade Mounting Medium | Preserves fluorescence signal by reducing photobleaching during imaging and storage. | ProLong Diamond, VECTASHIELD Antifade. |
| Multispectral or Confocal Imaging System | Capture high-resolution, multichannel images. Spectral systems enable unmixing of overlapping fluorophores. | Confocal microscopes (e.g., Zeiss LSM, Leica SP8), PhenoImagers (Akoya), Vectra systems. |
| Image Analysis Software | For quantitative analysis of marker expression, co-localization, and spatial relationships. | HALO, QuPath, ImageJ/FIJI, Imaris, inForm. |
1. Introduction: A Thesis Context This guide details the core experimental protocols for chromogenic (DAB) and fluorescent staining, providing the technical foundation for a comparative analysis within immunohistochemistry (IHC) and immunocytochemistry (ICC). The broader thesis investigates the critical trade-offs in sensitivity, multiplexing capability, spatial context, and quantification between these two primary detection methodologies in biomedical research and drug development.
2. The Scientist's Toolkit: Research Reagent Solutions
| Item | Primary Function in IHC/ICC |
|---|---|
| Primary Antibody | Binds specifically to the target antigen of interest. The core of assay specificity. |
| Chromogen (DAB/H2O2) | Enzyme substrate (for HRP) that yields an insoluble, brown precipitate upon oxidation. |
| Fluorophore Conjugate | Enzyme (HRP/AP) or fluorescent dye directly or indirectly attached to the detection antibody. |
| Blocking Serum | Reduces non-specific background staining by occupying unsaturated binding sites. |
| Antigen Retrieval Buffer | Unmasks epitopes altered by formalin fixation, critical for FFPE samples. |
| Mounting Medium | Aqueous (for fluorescence) or resin-based (for DAB); preserves signal and adds coverslip. |
| Nuclear Counterstain | Hematoxylin (for DAB) or DAPI/Hoechst (for fluorescence); provides tissue/cell architecture. |
| Autofluorescence Quencher | Reduces endogenous fluorescence in tissues (e.g., liver, kidney), improving signal-to-noise. |
3. Core Quantitative Comparison: DAB vs. Fluorescent Detection
Table 1: Key Characteristics of DAB and Fluorescent Detection Methods
| Parameter | Chromogenic (DAB) | Fluorescent |
|---|---|---|
| Signal Type | Permanent, insoluble precipitate | Emitted light (photons) |
| Detection Method | Brightfield microscopy | Epifluorescence/Confocal microscopy |
| Multiplexing | Sequential, limited (2-3plex max) | Simultaneous, high (4-8+ plex common) |
| Sensitivity | High (signal amplification via enzyme) | Very High (direct detection, low background) |
| Quantification | Semi-quantitative (density, intensity) | Highly Quantitative (linear range, pixel intensity) |
| Spatial Resolution | Excellent (with tissue context) | Superior (subcellular, confocal) |
| Permanence | High (slides archive for years) | Low (fluorophores bleach over time) |
| Primary Sample Type | FFPE tissues, high autofluorescence tissues | Cell cultures, frozen sections, multiplexed IHC |
4. Experimental Protocols
Protocol 4.1: Standard DAB Chromogenic IHC for FFPE Tissue Sections
Principle: HRP-conjugated secondary antibody catalyzes oxidation of DAB to form a brown precipitate at the antigen site.
- Dewaxing & Rehydration: Bake slides (60°C, 30 min). Deparaffinize in xylene (3 x 5 min) and rehydrate through graded ethanol (100%, 95%, 70% - 2 min each) to distilled water.
- Antigen Retrieval: Perform heat-induced epitope retrieval (HIER). Boil slides in 10mM Sodium Citrate buffer (pH 6.0) or Tris-EDTA (pH 9.0) for 20 min in a pressure cooker or steamer. Cool for 30 min at RT. Rinse in PBS.
- Endogenous Peroxidase Blocking: Incubate with 3% H2O2 in PBS (or methanol) for 10-15 min at RT to quench endogenous peroxidase activity. Wash in PBS.
- Blocking: Apply 2.5-5% normal serum (from species of secondary antibody) or protein block for 30 min at RT.
- Primary Antibody Incubation: Apply optimized dilution of primary antibody in antibody diluent. Incubate in a humidified chamber (1 hr at RT or overnight at 4°C). Wash in PBS-Tween (3 x 5 min).
- Secondary Antibody Incubation: Apply HRP-conjugated polymer secondary antibody (e.g., anti-mouse/rabbit EnVision system) for 30 min at RT. Wash in PBS (3 x 5 min).
- DAB Development: Prepare DAB substrate solution per manufacturer's instructions. Apply to tissue and monitor development under a microscope (typically 30 sec to 5 min). Stop reaction by immersing in distilled water.
- Counterstaining & Mounting: Counterstain with Hematoxylin (30 sec to 1 min), "blue" in tap water. Dehydrate through graded alcohols, clear in xylene, and mount with permanent resinous mounting medium.
Protocol 4.2: Standard Indirect Immunofluorescence (IF) for Cultured Cells (ICC)
Principle: Fluorophore-conjugated secondary antibody binds to primary antibody, enabling visualization via specific excitation/emission wavelengths.
- Fixation: Aspirate culture medium. Rinse cells gently with warm PBS. Fix with 4% paraformaldehyde in PBS for 15 min at RT. Alternative: Ice-cold methanol for 10 min at -20°C.
- Permeabilization & Blocking: (For intracellular targets) Incubate with 0.1-0.5% Triton X-100 in PBS for 10 min at RT. Wash with PBS (3 x 5 min). Apply blocking buffer (e.g., 5% BSA, 10% normal serum in PBS) for 1 hr at RT.
- Primary Antibody Incubation: Apply primary antibody diluted in blocking buffer. Incubate in a humidified chamber (1 hr at RT or overnight at 4°C). Wash with PBS (3 x 5 min).
- Secondary Antibody Incubation: Apply fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488, 555, 647) diluted in blocking buffer. Incubate for 1 hr at RT in the dark. Wash with PBS (3 x 5 min in the dark).
- Nuclear Counterstain & Mounting: Incubate with DAPI (300 nM in PBS) for 5 min at RT in the dark. Wash with PBS. Mount with an aqueous, anti-fade mounting medium (e.g., ProLong Gold). Seal coverslip with nail polish.
5. Visualized Workflows and Pathways
Diagram 1: DAB IHC Workflow for FFPE Tissue
Diagram 2: Fluorescent ICC Workflow for Cultured Cells
Diagram 3: Core Detection Principle: Indirect Method
This whitepaper provides an in-depth technical comparison of brightfield and fluorescence microscopy within the context of chromogenic versus fluorescent detection for immunohistochemistry (IHC) and immunocytochemistry (ICC). The choice of imaging modality is critical for data accuracy in research and drug development, directly impacting the quantification and localization of biomarkers.
Core Principles & Comparison
Brightfield Microscopy for Chromogenic Detection
Chromogenic detection utilizes enzymes such as horseradish peroxidase (HRP) or alkaline phosphatase (AP) to catalyze the deposition of a colored, precipitate-based chromogen (e.g., DAB, brown; AEC, red) at the antigen site. Imaging relies on transmitted white light, where the chromogen absorbs specific wavelengths, creating contrast against a counterstained background (e.g., hematoxylin).
Fluorescence Microscopy for Fluorescent Detection
Fluorescent detection employs fluorophore-conjugated antibodies or secondary reagents. Fluorophores absorb high-energy light (excitation) and emit lower-energy light (emission) at specific wavelengths. Imaging requires a set of filters: an excitation filter, a dichroic mirror, and an emission filter to isolate the signal from background.
Table 1: Quantitative Comparison of Imaging Modalities
| Parameter | Brightfield/Chromogenic | Fluorescence |
|---|---|---|
| Detection Limit | ~10⁶ molecules/μm² | ~10³ molecules/μm² |
| Dynamic Range | Low (~3-4 logs) | High (>5 logs) |
| Multiplexing Capacity | Typically 1-2 labels (color separation) | 4-8+ labels (spectral separation) |
| Spatial Resolution | ~250 nm (diffraction-limited) | ~200 nm (standard); <50 nm (super-res) |
| Signal Permanence | Stable, permanent stain | Prone to photobleaching |
| Background/Noise | High (endogenous pigments, scatter) | Low (optical sectioning possible) |
| Quantification Ease | Moderate (deconvolution needed) | High (direct signal measurement) |
| Typical Applications | Clinical pathology, morphology | Co-localization, live-cell, multiplex IHC/ICC |
Experimental Protocols for Comparative Analysis
Protocol: Multiplex Fluorescent IHC with Opal Polymer Detection
This protocol enables highly multiplexed biomarker analysis on a single FFPE tissue section.
- Deparaffinization & Antigen Retrieval: Bake slide at 60°C for 1 hr. Deparaffinize in xylene and rehydrate through graded ethanol to water. Perform heat-induced epitope retrieval (HIER) in citrate buffer (pH 6.0) or EDTA/Tris-EDTA buffer (pH 9.0) using a pressure cooker or steamer for 15-20 min.
- Primary Antibody Incubation: Block with Protein Block (e.g., 10% normal goat serum) for 10 min. Apply primary antibody (e.g., anti-CD3 rabbit monoclonal) diluted in antibody diluent. Incubate in a humidified chamber at room temperature for 1 hour or 4°C overnight.
- Polymer-HRP Secondary Incubation: Apply Opal Polymer HRP-conjugated secondary antibody (e.g., anti-rabbit) for 10 min at room temperature.
- Tyramide Signal Amplification (TSA): Apply Opal fluorophore reagent (e.g., Opal 520, 1:100 in amplification diluent) for 10 min. TSA deposition covalently binds the fluorophore to tyrosine residues near the antigen-antibody complex.
- Antibody Stripping: To remove the primary-secondary complex, heat slide in retrieval buffer again (HIER) for 20 min. This step denatures and elutes antibodies while leaving the deposited fluorophore intact.
- Repetition for Multiplexing: Repeat steps 2-5 for the next antibody-fluorophore pair (e.g., anti-CK, Opal 570; then anti-PD-L1, Opal 690).
- Counterstaining & Mounting: Stain nuclei with Spectral DAPI for 5 min. Mount with anti-fade mounting medium.
- Image Acquisition: Acquire using a multispectral fluorescence slide scanner or confocal microscope with defined filter sets for each fluorophore.
Protocol: Dual-Chromogenic IHC with Enzymatic Detection
This protocol is standard for visualizing two antigens with permanent stains.
- Deparaffinization & Retrieval: As per 3.1, Step 1.
- First Primary Antibody: Apply first mouse monoclonal primary antibody. Incubate.
- HRP Polymer & Chromogen: Apply anti-mouse HRP polymer. Visualize with DAB chromogen (produces a brown precipitate). Incubate for 5-10 min, then rinse.
- Antibody Blocking: To prevent cross-reactivity, apply a double stain block (e.g., from relevant kit) for 10 min. Alternatively, perform an acidic elution step (glycine-HCl buffer, pH 2.0, 10 min).
- Second Primary Antibody: Apply second rabbit monoclonal primary antibody. Incubate.
- AP Polymer & Chromogen: Apply anti-rabbit alkaline phosphatase (AP) polymer. Visualize with Fast Red/Vector Red (produces a red precipitate) or Vector Blue chromogen. Incubate for 10-15 min.
- Counterstaining & Mounting: Counterstain lightly with hematoxylin. Dehydrate through graded alcohols and xylene. Mount with permanent, non-aqueous mounting medium.
Slide Scanning & Digital Pathology Workflow
Whole-slide imaging (WSI) digitizes entire microscope slides. For brightfield, scanning typically uses a 20x or 40x objective with line-scan or area-scan cameras. For fluorescence, WSI requires motorized filter wheels, high-sensitivity cameras (e.g., sCMOS), and extended focus capabilities (z-stacking).
Key Scanning Parameters:
- Resolution: 0.25-0.5 μm/pixel for 40x equivalent.
- Focusing: Use tissue detection algorithms and multi-point autofocus.
- Fluorescence: Manage exposure time, illumination intensity (to limit photobleaching), and sequential channel capture.
Table 2: Analysis Software & Quantitative Outputs
| Analysis Type | Brightfield Chromogenic | Fluorescence Multiplex |
|---|---|---|
| Primary Software | HALO, QuPath, Indica Labs’ HALO AI | inForm, HALO, Visiopharm |
| Key Output Metrics | Positive cell count, H-Score, % area positivity, staining intensity (OD) | Cell phenotyping (positive/negative), density, co-expression analysis, spatial relationships (nearest neighbor) |
| Critical Step | Color deconvolution to separate chromogen (DAB) from counterstain (hematoxylin) | Spectral unmixing to separate overlapping fluorophore emissions |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Chromogenic vs. Fluorescent IHC/ICC
| Item | Function | Example Products/Types |
|---|---|---|
| Polymer-based Detection Systems | Amplifies signal by conjugating many enzymes (HRP/AP) to a polymer backbone linked to secondary antibody. Increases sensitivity over traditional methods. | ImmPress (Vector Labs), EnVision (Agilent), MACH (Biocare). |
| Tyramide Signal Amplification (TSA) Reagents | Enzymatic deposition of numerous fluorophore- or chromogen-labeled tyramide molecules, enabling ultra-sensitive detection and multiplexing. | Opal Polychromatic IHC Kits (Akoya), TSA Plus Kits (PerkinElmer). |
| Multiplex IHC/ICC Antibody Panels | Pre-validated sets of primary antibodies with confirmed compatibility for sequential staining protocols. | Cell Signaling Technology Multiplex IHC Panels, Abcam Multicellular IHC Panel. |
| Anti-fade Mounting Media | Preserves fluorescence signal by reducing photobleaching during imaging and storage. Contains radical scavengers. | ProLong Diamond (Thermo Fisher), Vectashield (Vector Labs). |
| Spectral DAPI | A nuclear counterstain with narrow emission, ideal for multiplex fluorescence as it minimizes bleed-through into other channels. | Spectral DAPI (Akoya), DAPI (Fluoroshield). |
| Multispectral Imaging Systems | Microscope or scanner capable of capturing the full emission spectrum at each pixel, enabling precise unmixing of overlapping fluorophores. | Vectra/Polaris (Akoya), PhenoImager (Akoya). |
| Automated Stainers | Provide consistent, reproducible staining for both chromogenic and fluorescent protocols, critical for high-throughput studies. | BOND RX (Leica), Autostainer (Agilent), LabSat (Akoya). |
Title: Chromogenic IHC Detection & Imaging Pathway
Title: Fluorescent IHC Detection & Imaging Pathway
Title: Sequential Multiplex Fluorescent IHC Workflow
The selection between brightfield/chromogenic and fluorescence detection is fundamental, dictated by research goals. Chromogenic IHC offers robust, permanent staining ideal for morphological context and clinical diagnostics. Fluorescent IHC/ICC provides superior sensitivity, multiplexing capacity, and quantifiability, essential for advanced research and drug development. The integration of these modalities with automated slide scanning and advanced image analysis pipelines forms the cornerstone of modern, quantitative tissue biomarker research.
This whitepaper details advanced applications of Immunocytochemistry (ICC) utilizing fluorescent detection, contextualized within a broader research thesis comparing chromogenic versus fluorescent detection in IHC/ICC. While chromogenic methods offer permanence and compatibility with brightfield microscopy, fluorescent detection is indispensable for modern, high-content, and dynamic analyses. This document focuses on three sophisticated, quantitative applications enabled by fluorescent ICC: co-localization analysis, live-cell imaging, and flow cytometry. These techniques provide unparalleled insights into protein interactions, cellular dynamics, and population heterogeneity, which are critical for drug discovery and mechanistic research.
Co-localization Studies: Quantitative Analysis of Protein Proximity
Co-localization analysis investigates the spatial overlap of two or more fluorescently labeled biomarkers within a cell, suggesting potential interaction or shared subcellular localization.
Key Methodologies & Quantitative Metrics
Quantitative co-localization moves beyond visual inspection. Current best practices employ statistical metrics, summarized in Table 1.
Table 1: Quantitative Co-localization Metrics and Their Interpretation
| Metric | Formula / Description | Interpretation | Ideal Value for True Co-localization |
|---|---|---|---|
| Pearson's Correlation Coefficient (PCC) | Measures linear dependence of intensity patterns: PCC = Σ[(Ri - R_avg)(Gi - G_avg)] / √[Σ(Ri - R_avg)² Σ(Gi - G_avg)²] |
Indicates correlation, not necessarily overlap. Insensitive to intensity changes. | +1 (perfect correlation). Values 0.5-1.0 suggest strong correlation. |
| Manders' Overlap Coefficients (M1 & M2) | Fraction of signal in one channel overlapping with the other: M1 = ΣRi,coloc / ΣRi ; M2 = ΣGi,coloc / ΣGi |
Measures actual signal overlap. Independent of intensity correlation. | 0 to 1. Values >0.5 indicate significant overlap. |
| Costes' Threshold | Iteratively sets intensity thresholds to calculate PCC. Validates if observed co-localization is above random chance. | Determines statistical significance. | PCC at threshold > 0, and >95% of randomized images yield lower PCC. |
Detailed Protocol: Confocal Microscopy for Co-localization
- Sample Preparation: Culture cells on #1.5 high-performance coverslips. Fix with 4% PFA for 15 min at RT. Permeabilize with 0.25% Triton X-100 for 10 min.
- Immunostaining: Block with 5% BSA/0.1% Tween-20 for 1 hour. Incubate with validated, host-specific primary antibodies (e.g., mouse anti-Protein A, rabbit anti-Protein B) overnight at 4°C. Use highly cross-adsorbed secondary antibodies conjugated to spectrally distinct fluorophores (e.g., Alexa Fluor 488 and Alexa Fluor 647).
- Image Acquisition: Acquire sequential (not simultaneous) images on a confocal microscope with high-resolution optics (63x/1.4 NA oil objective). Set pinhole to 1 Airy Unit. Use identical laser power, gain, and offset for all samples within an experiment.
- Analysis: Use specialized software (e.g., ImageJ with JACoP plugin, Imaris, Volocity). Apply background subtraction. Calculate PCC and M coefficients on thresholded images from minimum 30 cells per condition.
The Scientist's Toolkit: Co-localization
| Research Reagent / Material | Function & Critical Consideration |
|---|---|
| High-Performance Coverslips (#1.5H) | Ensure optical homogeneity and correct working distance for high-NA objectives. |
| Spectrally Distinct Fluorophores | Minimize bleed-through (e.g., Alexa Fluor 488 & 647 pair). Avoid overlapping emission spectra. |
| Cross-Adsorbed Secondary Antibodies | Eliminate species cross-reactivity, reducing false-positive co-localization signals. |
| Mounting Medium with Anti-fade | Presves fluorescence (e.g., with PPD or commercial reagents like ProLong Diamond). |
Title: Workflow for Quantitative Co-localization Analysis
Live-Cell Imaging ICC: Dynamics of Protein Localization
Live-cell ICC utilizes fluorescent proteins (FPs) or cell-permeable dyes/tags to monitor protein trafficking, organelle dynamics, and signaling events in real time.
Methodologies & Key Considerations
Table 2: Common Live-Cell ICC Approaches and Their Applications
| Approach | Mechanism | Typical Application | Temporal Resolution |
|---|---|---|---|
| Fluorescent Protein (FP) Fusions | Gene encoding protein of interest fused to GFP/RFP/etc. | Long-term tracking of protein expression, localization, and turnover. | Minutes to days. |
| Fluorescent Biosensors | FPs coupled to molecular switches (e.g., FRET-based). | Real-time detection of ions (Ca²⁺), phosphorylation, or enzymatic activity. | Seconds to minutes. |
| Cell-Permeable Fluorescent Dyes/Ligands | Small molecules targeting structures (e.g., MitoTracker, LysoTracker). | Labeling organelles or specific protein classes (e.g., HaloTag ligands). | Minutes to hours. |
| Fluorescent Nanobodies | Intracellularly expressed nanobodies binding to tags (e.g., GFP). | High-affinity tracking of endogenous or exogenous proteins. | Minutes to hours. |
Detailed Protocol: FRET-based Live-Cell Imaging for Kinase Activity
This protocol monitors kinase activity using a genetically encoded FRET biosensor.
- Biosensor Transfection: Transfect cells with plasmid encoding the FRET biosensor (e.g., AKAR family for PKA) using a method optimized for your cell line (e.g., lipofection, electroporation).
- Environmental Control: Plate cells on glass-bottom dishes. 24-48h post-transfection, place dish on a live-cell imaging system with controlled temperature (37°C), humidity, and CO₂ (5%).
- Image Acquisition: Use a widefield or confocal microscope capable of rapid, multi-channel acquisition. Excite the donor fluorophore (e.g., CFP at 433-456 nm). Collect emission from the donor channel (e.g., CFP: 465-510 nm) and the acceptor channel (e.g., YFP: 520-550 nm) simultaneously or sequentially with minimal delay.
- Stimulation & Data Collection: Acquire a 2-5 minute baseline. Add agonist (e.g., Forskolin for PKA) directly to dish without moving it. Continue time-lapse acquisition for 15-30 minutes.
- Analysis: Calculate the FRET ratio (Acceptor Emission / Donor Emission) for each time point after background subtraction. Plot ratio over time. Normalize to baseline (F/F₀).
The Scientist's Toolkit: Live-Cell Imaging
| Research Reagent / Material | Function & Critical Consideration |
|---|---|
| Glass-Bottom Culture Dish | Provides optimal optical clarity for high-resolution imaging. |
| Environment-Controlled Stage Top Chamber | Maintains cell viability by regulating temperature, CO₂, and humidity. |
| Low-Autofluorescence Phenol Red-Free Medium | Reduces background noise to enhance signal-to-noise ratio. |
| Genetically Encoded FRET Biosensor | Molecular tool that changes FRET efficiency upon a biochemical event (e.g., phosphorylation). |
Title: Mechanism of Live-Cell Imaging with a FRET Biosensor
Flow Cytometry ICC: High-Throughput Single-Cell Analysis
Flow cytometry applies fluorescent ICC to analyze antigen expression or modification across thousands to millions of individual cells, providing robust population statistics.
Methodologies & Data Output
Table 3: Key Outputs and Analysis from Flow Cytometry ICC
| Data Type | Description | Analytical Use |
|---|---|---|
| Median Fluorescence Intensity (MFI) | Median fluorescence of a cell population for a specific channel. | Measures relative antigen expression level. |
| Positive Cell Percentage (%) | Proportion of cells with fluorescence above a defined negative threshold. | Determines frequency of antigen-expressing cells. |
| Coefficient of Variation (CV) | Ratio of standard deviation to MFI. | Assesses staining uniformity or population heterogeneity. |
| Multiparameter Analysis | Correlative analysis of 2+ markers (e.g., bivariate dot plots). | Identifies and characterizes subpopulations (e.g., phenotyping). |
Detailed Protocol: Intracellular Phospho-Protein Staining for Flow Cytometry
This protocol detects phosphorylation states of signaling proteins (e.g., p-ERK, p-STAT) in single cells.
- Stimulation & Fixation: Stimulate cells in culture with target agonist for the desired time (e.g., 15 min). Immediately add an equal volume of pre-warmed 8% PFA directly to the culture medium to achieve 4% final concentration. Fix for 10-15 min at 37°C. Note: Fixation time and temperature are critical for preserving phospho-epitopes.
- Permeabilization: Pellet cells. Resuspend in ice-cold, 100% methanol. Vortex gently and incubate at -20°C for at least 30 minutes (or overnight). Methanol simultaneously permeabilizes membranes and extracts lipids, improving antibody access.
- Immunostaining: Wash cells twice in Flow Cytometry Staining Buffer (PBS with 1% BSA). Resuspend in staining buffer containing titrated, fluorophore-conjugated phospho-specific primary antibody. Incubate for 1 hour at RT in the dark. Direct conjugation avoids non-specific binding from secondary antibodies.
- Acquisition & Analysis: Wash cells and resuspend in buffer. Acquire data on a flow cytometer, collecting a minimum of 10,000 events per sample. Use an unstimulated, stained sample to set the negative population gate. Analyze MFI of the phospho-protein signal in the target population.
The Scientist's Toolkit: Flow Cytometry ICC
| Research Reagent / Material | Function & Critical Consideration |
|---|---|
| Rapid Fixation Solution (e.g., 8% PFA) | Instantly "freezes" transient phosphorylation states at the time of stimulation. |
| Methanol (100%, Ice-Cold) | Provides harsh permeabilization optimal for many nuclear and phospho-targets. |
| Flow Cytometry Staining Buffer | Protein-based buffer (BSA) blocks non-specific antibody binding to cells. |
| Directly Conjugated Phospho-Antibodies | Minimizes background and protocol steps; essential for multicolor panels. |
| Compensation Beads | Allow for precise spectral overlap correction in multicolor experiments. |
Title: Workflow for Intracellular Staining and Flow Cytometry ICC
Solving Common Problems: Optimization and Troubleshooting for Reliable Results
Within the ongoing research paradigm comparing chromogenic (DAB/BCIP/NBT) and fluorescent (FITC/Cy3/Alexa Fluor) detection for immunohistochemistry (IHC) and immunocytochemistry (ICC), a critical examination of chromogenic limitations is essential. This technical guide details the core pitfalls—background staining, poor contrast, and signal fading—offering methodological solutions grounded in current best practices to ensure data reproducibility and quantitative rigor.
Pitfall 1: High Background Staining
High background arises from non-specific antibody binding or endogenous enzyme activity, obscuring specific signal.
Pathophysiology & Mitigation Pathway
Key Experimental Protocol: Comprehensive Blocking
- Materials: Hydrogen peroxide (3%), levamisole (for alkaline phosphatase), normal serum from host species of secondary antibody, bovine serum albumin (BSA), Triton X-100 (for ICC).
- Procedure:
- Deparaffinize and rehydrate (FFPE sections).
- Quench endogenous peroxidases with 3% H₂O₂ in methanol for 10 min at RT.
- For AP-based systems, incubate with 1-2 mM levamisole for 30 min.
- Perform antigen retrieval if required (e.g., citrate buffer, pH 6.0, 95°C, 20 min).
- Apply protein block: Incubate with 2-5% normal serum + 1-3% BSA in PBS for 1 hour at RT.
- Proceed with primary antibody incubation.
Pitfall 2: Poor Optical Contrast
Weak or muddy contrast complicates image analysis and interpretation, often due to suboptimal chromogen precipitation or substrate exhaustion.
Quantitative Comparison of Chromogen Properties Table 1: Characteristics of Common Chromogenic Substrates
| Chromogen (Enzyme) | Precipitate Color | Recommended Counterstain | Relative Sensitivity | Fading Potential |
|---|---|---|---|---|
| DAB (HRP) | Brown | Hematoxylin | High | Low (Permanent) |
| AEC (HRP) | Red | Hematoxylin | Medium | High (Alcohol-soluble) |
| BCIP/NBT (AP) | Blue/Black | Nuclear Fast Red | Medium-High | Low |
| Vector VIP (HRP) | Purple | None/Methyl Green | High | Low |
Experimental Protocol: Enhanced Contrast Optimization
- Objective: Maximize signal-to-noise ratio for digital pathology.
- Method:
- Substrate Incubation Time: Perform kinetic monitoring. Incubate DAB for 30 seconds to 10 minutes; stop reaction at first sign of background (visualized under microscope).
- Enhanced Metallization (for DAB): Use commercial DAB enhancement kits (e.g., nickel or cobalt intensification) to shift precipitate to black/purple, increasing contrast against blue hematoxylin.
- Counterstain Selection: Use a light counterstain (e.g., diluted hematoxylin for DAB, Nuclear Fast Red for BCIP/NBT). Differentiate carefully.
- Dehydration & Mounting: For aqueous substrates (AEC), use aqueous mounting media. For DAB, use xylene and permanent resinous mounting medium.
Pitfall 3: Signal Fading Over Time
Chromogenic signals, while generally stable, can degrade due to photobleaching (certain substrates) or chemical interaction with mounting media.
Workflow for Ensuring Signal Permanence
Protocol: Archival-Grade Slide Preparation
- Post-Development Processing for DAB:
- After counterstaining, dehydrate slides through a graded ethanol series (70%, 95%, 100% x2), 2 minutes each.
- Clear in xylene or xylene substitute (3 changes, 2 minutes each).
- Mount under glass coverslips using a non-aqueous, DPX-type or entellan mounting medium.
- Digitization: Scan slides at high resolution immediately after curing (24-48 hours) to create a permanent digital record.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Mitigating Chromogenic Pitfalls
| Reagent | Primary Function | Key Consideration |
|---|---|---|
| Normal Serum (from secondary host) | Blocks non-specific protein-binding sites on tissue. | Must match the host species of the secondary antibody. |
| Endogenous Enzyme Block (H₂O₂, Levamisole) | Inactivates endogenous peroxidases or alkaline phosphatases. | Concentration and incubation time are critical to avoid tissue damage. |
| Polymer-based Detection System | Provides high sensitivity with low background vs. traditional avidin-biotin (ABC). | Streptavidin-free systems reduce non-specific binding to endogenous biotin. |
| Chromogen Kit (with Enhancer) | Provides optimized, stable substrate formulation (e.g., Metal-enhanced DAB). | Use freshly prepared working solution; monitor development kinetics. |
| Antigen Retrieval Buffer (Citrate/EDTA/TRIS) | Unmasks epitopes obscured by formalin fixation. | pH choice (6.0 vs 9.0) is antigen-dependent; requires optimization. |
| Aqueous Mounting Medium | Preserves alcohol-soluble chromogens (e.g., AEC). | Will dissolve DAB signal; use only for intended substrates. |
| Resinous Mounting Medium (DPX, Permount) | Provides permanent seal and optical clarity for dehydrated slides. | Requires complete dehydration; avoids bubbling over time. |
Addressing the core pitfalls of chromogenic detection—through rigorous blocking, contrast optimization, and archival-grade processing—ensures robust, quantifiable, and durable results. This methodological rigor is paramount for valid comparative analyses within the broader research thesis evaluating chromogenic versus fluorescent detection systems, directly impacting the reliability of data in translational research and drug development.
Within the broader analytical framework comparing chromogenic and fluorescent detection in IHC/ICC, fluorescent methods offer unparalleled multiplexing and quantification capabilities. However, their efficacy is critically limited by three persistent technical challenges: photobleaching, autofluorescence, and spectral overlap. This guide details current, practical strategies for their mitigation.
Photobleaching: Mechanisms and Mitigation
Photobleaching is the irreversible destruction of a fluorophore under illumination, diminishing signal over time. It involves complex photochemical pathways leading to permanent chemical modification.
Quantitative Comparison of Anti-Bleaching Reagents
Table 1: Efficacy of Common Photobleaching Mitigation Agents in Immunofluorescence
| Reagent / Strategy | Mechanism of Action | Typical Signal Retention Increase | Key Considerations |
|---|---|---|---|
| Commercial Anti-fade Mountants (e.g., ProLong Diamond) | Scavenge free radicals, reduce oxygen | 70-90% after 30 min illumination | Vary in refractive index; some harden. |
| p-Phenylenediamine (PPD) | Radical scavenging | ~60% after 20 min | Can be cytotoxic; may quench some dyes. |
| n-Propyl Gallate | Antioxidant | ~50% after 20 min | Use at low pH (e.g., in glycerol). |
| Trolox (vitamin E analog) | Stable antioxidant scavenger | Up to 80% after 30 min | Compatible with live-cell imaging. |
| Lower Illumination Intensity | Reduces photon flux | Variable, non-linear benefit | Requires more sensitive detectors (sCMOS, EM-CCD). |
| Oxygen Scavenging Systems (e.g., PCA/PCD) | Enzymatically removes oxygen | >90% in single-molecule assays | Complex to implement for standard IHC/ICC. |
Detailed Protocol: Trolox-Based Anti-Fade Mounting Medium
- Prepare a 10 mM Trolox stock solution in deionized water. Adjust pH to ~7.4, sterilize by filtration (0.2 µm), and store at 4°C protected from light for up to one week.
- For a 1x working solution, dilute the stock to a final concentration of 1-2 mM in your standard aqueous or glycerol-based mounting medium.
- After final washing of IHC/ICC samples, briefly drain excess buffer and mount slides with the Trolox-supplemented medium.
- Seal coverslip edges with clear nail polish or a commercial sealant. Store slides at 4°C in the dark.
Diagram Title: Photobleaching Pathways and Mitigation Strategies (100 chars)
Autofluorescence: Identification and Suppression
Autofluorescence (AF) is background emission from endogenous molecules (e.g., lipofuscin, NAD(P)H, collagen) upon excitation, obscuring specific signal.
Experimental Protocol: Chemical Quenching with Sudan Black B or TrueVIEW
Sudan Black B (SBB) Protocol:
- After completing all immunolabeling and final PBS washes, prepare a 0.1% (w/v) SBB solution in 70% ethanol.
- Incubate the mounted or unmounted tissue section/cells in the SBB solution for 10-20 minutes at room temperature, protected from light.
- Rinse thoroughly with PBS (3 x 5 min) to remove residual stain.
- Mount with antifade medium if not already mounted. SBB effectively quenches broad-spectrum AF, particularly from lipofuscin and red blood cells, without significantly affecting common synthetic fluorophores (e.g., Alexa Fluor dyes).
TrueVIEW Autofluorescence Quenching Kit Protocol:
- Follow standard IHC/ICC protocol through to the final wash before mounting.
- Prepare TrueVIEW reagent as per manufacturer's instructions.
- Apply reagent to the sample and incubate for the recommended time (typically 5-30 min).
- Rinse briefly with PBS or the provided buffer and proceed to mounting.
Spectral Overlap: Unmixing and Panel Design
Spectral overlap occurs when the emission tail of one fluorophore is detected in the channel of another, leading to crosstalk and false positives.
Quantitative Filter Set Selection
Table 2: Spectral Characteristics and Optimal Filter Sets for Common Fluorophores
| Fluorophore | Ex Max (nm) | Em Max (nm) | Recommended Dichroic (nm) | Key Overlap Considerations |
|---|---|---|---|---|
| DAPI / Hoechst | 359 | 457 | 405 | Minimal. Can bleed into FITC if filter is wide. |
| FITC / Alexa Fluor 488 | 495 | 519 | 495 | Significant bleed into Cy3/TRITC channel. |
| Cy3 / TRITC / Alexa Fluor 555 | 554 | 570 | 560 | Receives bleed-through from FITC. |
| Texas Red / Alexa Fluor 594 | 590 | 617 | 595 | Can bleed into Cy5 channel. |
| Cy5 / Alexa Fluor 647 | 650 | 665 | 660 | Minimal excitation by 488/568 nm lasers reduces crosstalk. |
| Dylight 405 | 400 | 420 | 405 | Can be imaged separately from DAPI with care. |
Detailed Protocol: Linear Unmixing for Multiplex Imaging
- Capture Single-Color Controls: For each fluorophore in your panel, prepare and image a control sample stained with that fluorophore alone, using the exact same acquisition settings (laser power, exposure time, filter sets) as for the multiplex experiment.
- Acquire Multiplex Image: Image the experimental sample containing all fluorophores.
- Generate Reference Spectra: For each control image, define a region of interest (ROI) over a brightly labeled area. Use your microscopy software's spectral unmixing tool to extract the emission spectrum for that fluorophore across all detection channels. This creates a reference spectrum library.
- Perform Unmixing: Apply the software's linear unmixing algorithm to the multiplex image. The algorithm uses the reference library to calculate the contribution of each fluorophore's unique spectrum to each pixel, thereby separating the signals mathematically.
Diagram Title: Spectral Unmixing Experimental Workflow (97 chars)
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Reagents for Mitigating Fluorescent Challenges
| Item | Category | Primary Function | Example/Brand |
|---|---|---|---|
| Phenylene-diamine (PPD) | Anti-fade Reagent | Radical scavenger; reduces photobleaching in fixed samples. | Sigma-Aldrich P6001 |
| ProLong Live/ Diamond | Anti-fade Mountant | Commercial formulations for live-cell or fixed-sample preservation. | Thermo Fisher Scientific |
| TrueVIEW Autofluorescence Quencher | Autofluorescence Suppression | Specifically reduces broad-spectrum tissue autofluorescence post-staining. | Vector Laboratories |
| Sudan Black B | Chemical Quencher | Lipophilic dye that quenches autofluorescence from lipids and lipofuscin. | Sigma-Aldrich 199664 |
| Cyclo-tetra-azacyclododecane (Cyclen) derivatives | Metal Chelation | Reduces aldehyde-induced autofluorescence by chelating metal ions. | Vector Laboratories REVEAL |
| Spectrally Matched Antibodies | Panel Design | Antibodies pre-conjugated to fluorophores with minimal overlap (e.g., Spark NIR). | BioLegend, Cytiva |
| Spectral Unmixing Software | Analysis Software | Enables computational separation of overlapping fluorophore signals. | ZEN (Zeiss), LAS X (Leica), INSPECTOR (Huygens) |
| sCMOS Camera | Detection Hardware | High quantum yield and sensitivity allow lower excitation light, reducing bleaching. | Hamamatsu Orca Fusion, Photometrics Prime |
Antibody and Dilution Optimization for Both Detection Types
Within the broader context of comparative research on chromogenic versus fluorescent detection in immunohistochemistry (IHC) and immunocytochemistry (ICC), antibody and dilution optimization emerges as the most critical foundational step. The choice of detection modality—chromogenic for brightfield microscopy or fluorescent for fluorescence microscopy—profoundly influences the optimization strategy, impacting parameters such as signal-to-noise ratio, multiplexing capability, and quantification potential. This guide provides a technical framework for systematically optimizing primary and secondary antibodies for both detection paradigms.
Foundational Principles: Chromogenic vs. Fluorescent Detection
The core difference lies in the signal generation. Chromogenic detection relies on an enzyme (typically Horseradish Peroxidase or Alkaline Phosphatase) conjugated to a secondary antibody to catalyze the precipitation of a colored dye (e.g., DAB, Vector Red) at the antigen site. Fluorescent detection uses a fluorophore (e.g., Alexa Fluor dyes, Cy dyes) conjugated to a secondary antibody, which emits light of a specific wavelength upon excitation.
Key Optimization Considerations:
- Chromogenic: Signal amplification is high due to enzyme-substrate kinetics, but dynamic range is limited (yes/no signal, saturation). Optimization focuses on maximizing specific precipitate while minimizing endogenous enzyme activity and non-specific background. Multiplexing is limited to 2-3 targets using different enzymes.
- Fluorescent: Signal is linear over a wider range, enabling better quantification. Optimization focuses on maximizing specific fluorescence while minimizing autofluorescence and bleed-through between channels. Enables high-plex multiplexing (4+ targets).
Systematic Optimization Workflow
Diagram Title: Antibody Optimization Core Workflow
Phase 1: Primary Antibody Titration (Checkerboard Assay)
The goal is to identify the highest dilution that yields a strong specific signal with minimal background.
Protocol:
- Prepare serial dilutions of the primary antibody (e.g., 1:50, 1:100, 1:200, 1:500, 1:1000) in an appropriate antibody diluent.
- Apply these dilutions to serial tissue/cell sections or wells containing the same biological sample.
- For each primary antibody dilution, also titrate the detection system component (secondary antibody or full detection kit). This creates a 2D "checkerboard."
- Develop with chromogenic substrate or mount for fluorescence.
- Score results based on Signal Intensity and Background Staining (See Table 1).
Scoring Criteria:
- Signal Intensity: 0 (No signal) to 4+ (Very strong signal).
- Background: 0 (None) to 3+ (High, obscures specific signal).
- Optimal Point: Highest signal intensity (3+ or 4+) with the lowest background (0 or 1+) at the highest primary antibody dilution.
Table 1: Example Checkerboard Results for a Hypothetical Target
| Primary Ab Dilution | Secondary/Detector Dilution | Chromogenic Signal (DAB) | Chromogenic Background | Fluorescent Signal (Cy3) | Fluorescent Background |
|---|---|---|---|---|---|
| 1:50 | 1:100 (Ready-to-Use) | 4+ | 3+ (High) | 4+ | 3+ (High) |
| 1:100 | 1:200 | 4+ | 2+ (Moderate) | 4+ | 2+ |
| 1:200 | 1:400 | 4+ | 1+ (Low) | 4+ | 1+ |
| 1:500 | 1:400 | 2+ (Weak) | 0 | 3+ | 0 |
| 1:1000 | 1:400 | 1+ | 0 | 2+ | 0 |
Phase 2: Detection System Optimization
This phase is distinct for each detection type.
Chromogenic Detection Protocol (Indirect HRP):
- After primary antibody incubation, apply enzyme-conjugated secondary antibody (e.g., HRP-anti-rabbit) at the optimized dilution for 30-60 minutes.
- Prepare chromogen substrate (e.g., DAB). Critical: Optimize incubation time (typically 30 seconds to 10 minutes) under microscope monitoring to prevent over-development and increased background.
- Stop reaction in water. Apply hematoxylin counterstain, differentiate, and mount with aqueous mounting medium.
Fluorescent Detection Protocol (Indirect):
- After primary antibody incubation, apply fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488-anti-mouse) at the optimized dilution for 30-60 minutes in the dark.
- Apply nuclear counterstain compatible with fixation (e.g., DAPI, Hoechst).
- Mount with a fluorescent anti-fade mounting medium (e.g., ProLong Diamond).
Diagram Title: Core Detection Signaling Pathways
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function & Importance in Optimization |
|---|---|
| Validated Primary Antibodies | Specificity is paramount. Use antibodies validated for IHC/ICC in the relevant species. Knockout/knockdown tissue controls are ideal. |
| Antibody Diluent | A buffered protein solution (e.g., with BSA or serum) that stabilizes antibodies and reduces non-specific binding to tissue. |
| Polymer-based Detection Systems | Pre-formed polymers with multiple secondary antibodies and enzymes/fluorophores. Offer superior sensitivity and lower background than traditional methods for both chromogenic and fluorescent detection. |
| Fluorophore Conjugates (e.g., Alexa Fluor, Cy dyes) | Bright, photostable dyes with distinct emission spectra. Choice depends on microscope filter sets and multiplexing panel design. |
| Chromogen Substrates (e.g., DAB, Vector Red, BCIP/NBT) | Enzyme substrates that yield insoluble precipitates. DAB is most common (brown), but others offer different colors for multiplexing. |
| Fluorophore-Antifade Mounting Media | Preserves fluorescence intensity by reducing photobleaching. Critical for imaging and archival of fluorescent samples. |
| Automated Staining Platforms | Provide exceptional reproducibility in reagent application, incubation times, and washing, essential for standardized optimization and high-throughput work. |
Table 2: Direct Comparison of Optimization Parameters
| Parameter | Chromogenic Detection | Fluorescent Detection |
|---|---|---|
| Key Primary Ab Dilution Goal | Avoid excess antibody that increases background precipitate. Often used at lower dilutions (e.g., 1:100-1:200). | Can often be used at higher dilutions (e.g., 1:500-1:1000) as signal is directly proportional to fluorophore. |
| Signal Amplification | High (enzymatic deposition). | None (1º Ab : Fluorophore = ~1:1 to 1:several with polymers). |
| Critical Variable to Titrate | Chromogen incubation time. Directly controls signal intensity/background. | Fluorophore concentration & imaging exposure time. Controls signal intensity and channel bleed-through. |
| Background Sources | Endogenous enzyme activity, non-specific antibody binding, over-development. | Autofluorescence, non-specific antibody binding, spectral bleed-through. |
| Multiplexing Approach | Sequential staining with different enzymes (HRP, AP), heat-mediated antibody stripping. | Simultaneous staining with spectrally distinct fluorophores. |
| Optimal Counterstain | Hematoxylin (nuclear, blue). | DAPI, Hoechst (nuclear, blue), or SYTOX dyes. |
| Mounting Medium | Aqueous, permanent. | Anti-fade, non-aqueous. |
Final Validation Protocol for Chromogenic IHC (Optimized for DAB/HRP):
- Deparaffinize and rehydrate FFPE tissue sections.
- Perform heat-induced epitope retrieval in citrate buffer (pH 6.0) for 20 min.
- Block endogenous peroxidase with 3% H₂O₂ for 10 min.
- Block non-specific protein binding with 2.5% normal horse serum for 20 min.
- Incubate with optimized primary antibody dilution (from checkerboard) overnight at 4°C.
- Apply HRP-polymer conjugate secondary antibody (e.g., ImmPRESS) for 30 min at RT.
- Develop with DAB substrate for precisely 90 seconds (optimized time). Monitor slides microscopically.
- Counterstain with hematoxylin for 30 seconds, differentiate, blue.
- Dehydrate, clear, and mount with permanent mounting medium.
Final Validation Protocol for Fluorescent ICC (Optimized for Multiplexing):
- Culture and fix cells with 4% PFA for 15 min at RT. Permeabilize with 0.1% Triton X-100.
- Block with 5% BSA / 0.1% Tween-20 for 1 hour.
- Incubate with cocktail of optimally diluted primary antibodies raised in different hosts (e.g., mouse anti-target A, rabbit anti-target B) overnight at 4°C.
- Apply cocktail of spectrally distinct, cross-adsorbed secondary antibodies (e.g., donkey anti-mouse AF488, donkey anti-rabbit AF555) for 1 hour at RT in the dark.
- Counterstain nuclei with DAPI (300 nM) for 5 min.
- Mount with ProLong Diamond Antifade Mountant. Cure for 24 hours before imaging.
- Image using sequential acquisition to minimize bleed-through.
Sample Preparation and Fixation Considerations for Optimal Signal
Within the comprehensive investigation of chromogenic versus fluorescent detection in immunohistochemistry (IHC) and immunocytochemistry (ICC), sample preparation and fixation constitute the foundational, non-negotiable steps that dictate the success or failure of any subsequent signal generation. The choice between chromogenic and fluorescent detection is often secondary to the quality of antigen preservation and tissue morphology achieved during initial processing. This guide details the critical technical parameters of sample preparation and fixation, framed by their direct impact on signal optimization for both detection paradigms.
The Critical Role of Fixation
Fixation halts degradation, preserves morphological structure, and immobilizes antigens. The method and duration directly influence epitope accessibility, background signal, and autofluorescence—factors with divergent implications for chromogenic and fluorescent IHC/ICC.
Common Fixatives: Mechanisms and Impacts
Formalin (10% Neutral Buffered Formalin, NBF):
- Mechanism: Crosslinks proteins via methylene bridges.
- Effect on Signal: Excellent morphology but can mask epitopes, often necessitating antigen retrieval. Can introduce autofluorescence (broad emission ~430-540 nm), which is problematic for fluorescent detection but irrelevant for chromogenic.
Paraformaldehyde (PFA, 2-4%):
- Mechanism: Similar crosslinking to NBF but purer and faster penetrating.
- Effect on Signal: Preferred for cell-based ICC and delicate tissues. Controlled fixation time is crucial to balance preservation with over-fixation.
Acetone/Methanol:
- Mechanism: Precipitates proteins (denaturation).
- Effect on Signal: Excellent for preserving many epitopes, especially phosphorylated sites. No crosslinking, so antigen retrieval is not required. Methanol fixation reduces autofluorescence compared to aldehydes. Can compromise fine cellular morphology.
Table 1: Quantitative Comparison of Fixative Effects on Signal Parameters
| Fixative Type | Optimal Concentration & Time | Primary Impact on Chromogenic IHC | Primary Impact on Fluorescent IHC/ICC | Key Consideration |
|---|---|---|---|---|
| NBF | 10%, 18-24 hrs (tissue) | May reduce final DAB/HRP signal intensity; requires AR. | Increases risk of autofluorescence; requires AR and quenching. | Over-fixation necessitates harsher AR, damaging tissue. |
| PFA | 4%, 10-30 min (cells), 6-24 hrs (tissue) | Consistent, strong signal post-AR. Can be milder than NBF. | Autofluorescence present but manageable. Optimal for multiplexed ICC. | Time is critical; longer fixation increases crosslinking. |
| Acetone | 100%, -20°C, 10 min | High signal for many antibodies without AR. Poor morphological detail. | Low autofluorescence. Ideal for surface antigens in ICC. | Destroys fine structure; not suitable for all tissues. |
| Methanol | 100%, -20°C, 10 min | Similar to acetone. Can be combined with acetone. | Low autofluorescence. Permeabilizes simultaneously. | Causes cell shrinkage and precipitation artifacts. |
Optimized Experimental Protocols
Protocol A: Standard Tissue Fixation and Processing for Chromogenic IHC
Objective: Preserve morphology and antigenicity for brightfield, chromogenic detection.
- Dissection & Fixation: Immerse tissue in 10% NBF within 1 minute of excision. Fix for 18-24 hours at 4°C (volume 20x tissue volume).
- Washing: Rinse in 1X PBS, pH 7.4, for 24 hours with 3-4 buffer changes to remove excess fixative.
- Dehydration & Clearing: Process tissue through a graded ethanol series: 70% EtOH (2 hrs), 95% EtOH (2 x 1 hr), 100% EtOH (2 x 1 hr). Clear in xylene or xylene-substitute (2 x 1 hr).
- Infiltration & Embedding: Infiltrate with molten paraffin wax at 55-60°C (2-3 changes, 1 hr each). Embed in fresh paraffin blocks.
- Sectioning: Cut 4-5 µm sections using a microtome. Float on a 42°C water bath. Mount on charged or adhesive glass slides.
- Drying: Dry slides overnight at 37°C or for 1 hour at 60°C to ensure adhesion.
Protocol B: Cell Culture Fixation & Permeabilization for Multiplex Fluorescent ICC
Objective: Maximize epitope retention, minimize autofluorescence, and allow antibody penetration for multiplexed, fluorescent detection.
- Culture: Grow cells on #1.5 coverslips in a multi-well plate.
- Rinsing: Aspirate media. Gently rinse cells 2x with pre-warmed (37°C) 1X PBS, pH 7.4.
- Fixation: Add 4% PFA in PBS to cover cells. Fix for 15 minutes at room temperature (RT).
- Washing: Wash cells 3x with PBS (5 min per wash).
- Permeabilization/Quenching (Simultaneous): Incubate cells in a freshly prepared solution of 0.1% Triton X-100 + 0.1 M Glycine in PBS for 15 minutes at RT. (Glycine quenches unreacted aldehydes, reducing autofluorescence).
- Blocking: Incubate cells in blocking buffer (e.g., 5% BSA, 0.05% Tween-20 in PBS) for 1 hour at RT.
- Proceed to Immunostaining: Dilute primary antibodies in antibody dilution buffer (e.g., 1% BSA in PBS).
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Sample Preparation
| Item | Function in Preparation/Fixation | Key Consideration |
|---|---|---|
| Neutral Buffered Formalin (10% NBF) | Standard tissue fixative; crosslinks proteins for morphology. | Must be fresh (<1 year old); pH should be 7.0-7.4. |
| Paraformaldehyde (PFA), 16-32% Stocks | Gold-standard for cellular fixation; purer than NBF. | Aliquot and store at -20°C; avoid freeze-thaw. Make 4% dilution fresh. |
| Phosphate-Buffered Saline (PBS), 10X | Isotonic washing and dilution buffer. | Use at 1X, pH 7.4. Calcium/Magnesium-free is standard. |
| Glycine Solution (1M Stock) | Quenches unreacted aldehyde groups, reducing background and autofluorescence. | Critical step post-PFA/NBF fixation for fluorescence. |
| Triton X-100 or Tween-20 | Non-ionic detergent for permeabilizing cell membranes. | Concentration (0.1-0.5%) and time must be optimized per antigen. |
| Bovine Serum Albumin (BSA), Fraction V | Component of blocking and antibody dilution buffers; reduces non-specific binding. | Use at 1-5% in PBS or TBS. |
| Sodium Borohydride (NaBH4) | Strong reducing agent for quenching autofluorescence in aldehyde-fixed tissues. | Use with caution (0.1% in PBS, 10 min). Can damage some epitopes. |
| Cryoprotectant (e.g., 30% Sucrose) | For frozen sections; prevents ice crystal formation that destroys morphology. | Infiltrate fixed tissue until it sinks (overnight at 4°C) before OCT embedding. |
Visualizing the Workflow and Impact
Workflow: Fixation Path to Detection Choice
Fixation Factors Impacting Final Signal
Within the critical research context of comparing chromogenic versus fluorescent detection for immunohistochemistry (IHC) and immunocytochemistry (ICC), signal amplification is paramount. It directly impacts assay sensitivity, multiplexing capability, and quantification accuracy. This guide details the core principles, protocols, and best practices for two dominant amplification methodologies: Tyramide Signal Amplification (TSA, also known as CARD) and labeled polymer systems.
Fundamental Principles & Comparative Framework
Tyramide Signal Amplification (TSA)
TSA is an enzyme-mediated deposition technique. A peroxidase enzyme (typically HRP) conjugated to a primary or secondary antibody catalyzes the conversion of tyramide substrates into highly reactive intermediates. These intermediates covalently bind to electron-rich residues (e.g., tyrosine) on proteins proximal to the enzyme, depositing numerous fluorophores or haptens per target. This enables extreme signal amplification and is exceptionally suited for fluorescent multiplexing.
Labeled Polymer Systems
These systems (e.g., EnVision, ImmPRESS) involve a dextran or other polymer backbone directly conjugated to a large number of enzyme molecules (HRP or AP) and secondary antibodies. The polymer binds to the primary antibody, delivering a high enzyme-to-target ratio in a single step. This method enhances sensitivity over standard streptavidin-biotin methods and reduces background.
Table 1: Core Comparison of Amplification Systems
| Feature | Tyramide Signal Amplification (TSA) | Labeled Polymer Systems |
|---|---|---|
| Amplification Mechanism | Enzyme-catalyzed deposition of labels | Pre-formed polymer with multiple enzymes |
| Typical Gain | 100-1000x over direct methods | 10-100x over standard indirect methods |
| Best Suited For | High-plex fluorescence IHC/ICC, low-abundance targets | High-sensitivity single-plex chromogenic IHC |
| Multiplexing Flexibility | Very High (sequential rounds with HRP inactivation) | Low (typically single label per round) |
| Spatial Resolution | Very High (deposition is localized) | High (polymer is confined to target site) |
| Background Risk | Moderate (requires precise optimization) | Low (no endogenous biotin interference) |
| Protocol Complexity | High | Low (often a one-step post-primary) |
Detailed Experimental Protocols
Protocol for Fluorescent TSA Multiplexing
This protocol is for sequential detection of three antigens on formalin-fixed, paraffin-embedded (FFPE) tissue.
Key Reagents & Solutions:
- TSA Reagent Kits: Fluorophore-conjugated tyramides (e.g., Cy3, Cy5, FITC).
- Peroxidase Block: 3% H₂O₂ in methanol.
- Antigen Retrieval Buffer: Citrate, pH 6.0, or EDTA/TRIS, pH 9.0.
- Blocking Buffer: Protein block (serum or BSA).
- Primary Antibodies: Host species must be identical OR require species-specific polymer systems for some rounds.
- HRP Polymer Conjugate: Anti-host IgG polymer (e.g., anti-rabbit HRP).
- Antibody Elution Buffer: Glycine-HCl, pH 2.0, or SDS-based buffer.
Methodology:
- Deparaffinization & Antigen Retrieval: Perform standard dewaxing and heat-induced epitope retrieval.
- Peroxidase Blocking: Incubate with 3% H₂O₂ for 15 min to quench endogenous peroxidase.
- Blocking: Apply protein block for 30 min.
- Primary Antibody 1: Incubate with first mouse monoclonal antibody (1 hr, RT or overnight, 4°C).
- Polymer Amplification: Incubate with anti-mouse HRP polymer for 30 min.
- Tyramide Deposition: Incubate with first fluorophore-tyramide (1:100 dilution in provided amplifier) for 5-10 min.
- HRP Inactivation: Treat slides with 3% H₂O₂ for 15 min to inactivate HRP from round 1.
- Antibody Elution (Optional but Recommended): Incubate in heated antigen retrieval buffer or glycine buffer (pH 2.0) for 10-20 min to remove primary/secondary complexes.
- Repeat Cycle (Steps 2-7): For antibody 2 (e.g., rabbit polyclonal), using anti-rabbit HRP polymer and a different fluorophore-tyramide. Omit global peroxidase block in step 2 of subsequent cycles.
- Repeat for Antibody 3.
- Counterstain & Mount: Apply DAPI and mount with anti-fade medium.
Protocol for Polymer-Based Chromogenic Detection
This protocol is optimized for single-plex, high-contrast chromogenic detection.
Key Reagents & Solutions:
- Polymer-HRP Conjugate: One-step anti-host Ig polymer (e.g., ImmPRESS HRP Anti-Rabbit IgG).
- Chromogen Substrate: DAB (3,3'-Diaminobenzidine) or AEC (3-Amino-9-ethylcarbazole).
- Hematoxylin Counterstain.
Methodology:
- Deparaffinization, Antigen Retrieval, and Peroxidase Blocking as in 3.1.
- Blocking: Apply appropriate serum block for 20 min.
- Primary Antibody Incubation: Apply rabbit primary antibody for 1 hr.
- Polymer Conjugate Incubation: Apply anti-rabbit HRP polymer conjugate for 30 min. No tertiary step required.
- Chromogen Development: Incubate with prepared DAB substrate for 3-10 min; monitor under microscope.
- Counterstain: Immerse in hematoxylin for 30-60 sec.
- Dehydration & Mounting: Dehydrate through graded alcohols, clear in xylene, and mount with permanent resinous medium.
Visualization of Workflows & Pathways
Title: Comparative Workflow of TSA vs Polymer Amplification
Title: TSA Biochemical Pathway Mechanism
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Signal Amplification Experiments
| Reagent Category | Specific Example | Function & Critical Note |
|---|---|---|
| Amplification System | Opal TSA Fluorophore Kits | Ready-to-use tyramide reagents for multiplex fluorescence. Critical: Each fluorotype requires individual optimization. |
| Polymer Conjugate | ImmPRESS HRP Polymer | Species-specific polymer for chromogenic IHC. Critical: Eliminates non-specific biotin background. |
| Enzyme Substrate | DAB Chromogen Kit | Yields brown, alcohol-insoluble precipitate. Critical: Requires careful timing and disposal as a potential carcinogen. |
| Blocking Solution | Normal Serum (e.g., Goat) | Reduces non-specific binding of secondary antibodies. Critical: Must match the species of the polymer/secondary antibody. |
| Peroxidase Block | 3% Aqueous H₂O₂ | Inactivates endogenous tissue peroxidase. Critical: Essential for low-background chromogenic and TSA assays. |
| Antibody Elution Buffer | pH 2.0 Glycine-HCl or SDS | Removes primary/secondary complexes between TSA rounds. Critical: Validation required to ensure antigen integrity is maintained. |
| Mounting Medium | ProLong Diamond Antifade | For fluorescent samples; preserves fluorescence. Critical: Use aqueous-based (e.g., Fluoromount) for AEC chromogen. |
Side-by-Side Analysis: Validating and Comparing Detection Methods for Robust Data
This technical guide provides a detailed comparison between 3,3'-Diaminobenzidine (DAB)-based chromogenic and fluorescent detection methods within the broader research context of comparing chromogenic versus fluorescent detection for immunohistochemistry (IHC) and immunocytochemistry (ICC). The selection of detection system is fundamental, impacting sensitivity, multiplexing capability, and compatibility with downstream analyses.
Quantitative Comparison of Core Attributes
Table 1: Performance and Practicality Metrics
| Attribute | DAB (Chromogenic) | Fluorescent Detection |
|---|---|---|
| Sensitivity | High (signal amplification via enzyme precipitation). | Very High to Extreme (e.g., Tyramide Signal Amplification (TSA) can detect single molecules). |
| Detection Limit (Approx.) | ~100s of copies per cell. | Can be <10 copies per cell with amplification. |
| Spatial Resolution | Excellent, precise localization due to precipitate. | Excellent, limited by diffraction of light (~200 nm). |
| Signal Stability | Permanent, does not photobleach. | Temporary, susceptible to photobleaching; requires antifade mounting. |
| Multiplexing Capacity | Low (typically 1-2 markers on serial sections). | High (4-8+ markers simultaneously via spectral separation). |
| Background (Typical) | Lower endogenous background in many tissues. | Higher potential for tissue autofluorescence. |
| Compatibility w/ Brightfield | Yes, standard for pathology. | No, requires fluorescence/confocal microscopy. |
| Quantification Method | Densitometry (intensity of brown precipitate). | Fluorescence intensity (linear over a wider range). |
| Cost per Assay | Generally lower. | Higher (antibodies, mounting media, imaging systems). |
Table 2: Application-Specific Suitability
| Application Context | Recommended Method | Primary Rationale |
|---|---|---|
| Clinical Diagnostics / Pathology | DAB | Permanent slides, standard brightfield viewing, familiar interpretation. |
| High-plex Spatial Phenotyping | Fluorescent | Ability to label multiple antigens in a single tissue section. |
| Co-localization Studies | Fluorescent | Precise pixel-level overlap analysis of two+ targets. |
| Low-Abundance Target Detection | Fluorescent (with TSA) | Superior sensitivity with amplification. |
| Combination with In Situ Hybridization | Either | DAB for brightfield FISH; fluorescent for multiplex RNA/protein. |
| Archival Tissue (Long-term storage) | DAB | Stable signal over decades. |
Experimental Protocols
Protocol 1: Standard DAB Chromogenic IHC
- Deparaffinization & Rehydration: Process formalin-fixed, paraffin-embedded (FFPE) sections through xylene and graded ethanol series to water.
- Antigen Retrieval: Heat slides in citrate buffer (pH 6.0) or EDTA/Tris-EDTA buffer (pH 9.0) using a pressure cooker or microwave.
- Endogenous Peroxidase Blocking: Incubate with 3% hydrogen peroxide for 10 minutes.
- Blocking: Apply serum-free protein block for 10 minutes to reduce non-specific binding.
- Primary Antibody Incubation: Apply species-specific, validated primary antibody; incubate at 4°C overnight or at room temperature for 1 hour.
- Secondary Antibody Incubation: Apply horseradish peroxidase (HRP)-conjugated polymer secondary antibody for 30 minutes.
- DAB Development: Apply prepared DAB chromogen substrate (e.g., 1 drop DAB chromogen to 1 ml substrate buffer). Monitor development under a microscope (typically 30 seconds to 5 minutes). Stop reaction in water.
- Counterstaining: Immerse in hematoxylin for 30-60 seconds, then blue in running tap water.
- Dehydration & Mounting: Dehydrate through graded ethanol, clear in xylene, and mount with a permanent mounting medium.
Protocol 2: Standard Multiplex Fluorescent IHC/ICC
- Sample Preparation: Fix cells/tissue (e.g., 4% PFA for 15 min). Permeabilize with 0.1% Triton X-100 (for intracellular targets).
- Autofluorescence & Non-Specific Blocking: Incubate with a blocking buffer (e.g., 5% normal serum, 3% BSA) containing a reducing agent (e.g., 0.1% sodium borohydride) if needed.
- Primary Antibody Incubation: Apply primary antibody from host species A, incubate as per validation.
- Fluorescent Secondary/Amplification: Apply fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488) or use a TSA kit (e.g., Opal) for amplified signal. Incubate protected from light.
- Antibody Stripping (for Sequential Labeling): For multiplexing with same-species primaries, apply a mild stripping agent (e.g., heat in pH 6.0 buffer) to remove antibody complexes without damaging antigens.
- Repeat Steps 3-5: For subsequent targets, using different fluorophores (e.g., Cy3, Cy5).
- Counterstaining & Mounting: Apply nuclear counterstain (e.g., DAPI, 1 µg/mL for 5 min). Mount with antifade mounting medium (e.g., ProLong Diamond).
- Imaging: Image using a fluorescence or confocal microscope with appropriate filter sets. Acquire and merge channels digitally.
Visualization of Key Methodologies
Title: DAB vs Direct Fluorescent Detection Workflows
Title: Tyramide Signal Amplification (TSA) Principle
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Research Reagents and Materials
| Item | Primary Function & Rationale |
|---|---|
| Polymer-HRP Secondary | High-sensitivity, low-background conjugate for DAB detection; avoids biotin/avidin systems. |
| Ready-to-Use DAB Kit | Stable, consistent chromogen/substrate mix; contains essential inhibitors for controlled development. |
| Fluorophore-Conjugated Secondary | Highly cross-adsorbed antibodies with bright, photostable dyes (e.g., Alexa Fluor series) for multiplexing. |
| Tyramide Signal Amplification (TSA) Kit | Provides enzyme substrates for radical generation, enabling extreme signal amplification for low-copy targets. |
| Antibody Stripping Buffer | Allows sequential labeling with same-species primary antibodies in multiplex fluorescent IHC. |
| Antifade Mounting Medium | Preserves fluorophore signal by reducing photobleaching; often contains DAPI for nuclear counterstain. |
| Multispectral Imaging System | For fluorescent multiplexing, enables spectral unmixing to separate overlapping emission spectra and eliminate autofluorescence. |
| Automated Image Analysis Software | Enables quantitative analysis of DAB stain intensity (positive pixel count) and fluorescent co-localization (e.g., Manders' coefficients). |
Within the continuum of chromogenic (DAB) versus fluorescent detection for immunohistochemistry (IHC) and immunocytochemistry (ICC), the selection of an optimal quantitative analysis method is a critical determinant of research validity. This guide provides a technical comparison of mainstream quantification methodologies, contextualized by their application to each detection paradigm, to inform biomarker scoring in research and therapeutic development.
Core Quantitative Methodologies: Principles and Applications
Quantitative image analysis transforms pixel data into objective, reproducible biomarker metrics. The suitability of a method is intrinsically linked to the detection chemistry.
Brightfield (Chromogenic) Image Analysis
Chromogenic detection, typically with 3,3'-Diaminobenzidine (DAB), produces a localized brown precipitate. Quantification relies on color deconvolution and optical density measurement.
- Color Deconvolution: Separates the DAB (brown) signal from the hematoxylin (blue) counterstain using fixed or adaptive optical density vectors.
- Optical Density (OD) Measurement: The OD of the DAB precipitate is theoretically proportional to the amount of target antigen, following the Beer-Lambert law. Analysis yields metrics like H-Score and DAB Pixel Intensity.
Fluorescence Image Analysis
Fluorescent detection uses fluorophore-conjugated antibodies emitting light at specific wavelengths upon excitation. Quantification is based on fluorescence intensity within defined regions.
- Spectral Unmixing: Separates overlapping emission spectra from multiple fluorophores.
- Intensity Measurement: Direct measurement of pixel intensity values, corrected for background autofluorescence. Common outputs include Mean Fluorescence Intensity (MFI), Total Fluorescence, and Co-localization Coefficients.
Table 1: Quantitative Method Comparison for Chromogenic vs. Fluorescent IHC/ICC
| Parameter | Chromogenic (DAB) Quantification | Fluorescent Quantification |
|---|---|---|
| Primary Metric | Optical Density (OD), Positive Pixel Area | Mean Fluorescence Intensity (MFI) |
| Key Scoring Algorithms | H-Score, Allred Score, QuPath Positivity % | MFI Ratio, % Positive Cells (by intensity threshold), Co-localization (Manders'/Pearson's) |
| Linear Dynamic Range | Narrow (subject to enzyme saturation) | Broad (dependent on detector saturation) |
| Multiplexing Capability | Low (typically 1-2 markers + counterstain) | High (4+ markers with spectral unmixing) |
| Sensitivity to Background | High (affected by endogenous pigments, staining artifacts) | Moderate (requires robust autofluorescence correction) |
| Tissue Morphology Context | Excellent (brightfield enables clear H&E-like visualization) | Challenging (often requires separate brightfield or nuclear stain channel) |
| Common Analysis Platforms | Aperio ImageScope, QuPath, HALO, Indica Labs Halo | ImageJ/FIJI, CellProfiler, Visiopharm, Leica LAS X |
| Best Suited For | High-throughput clinical pathology scoring, single biomarker with clear subcellular localization. | Research multiplexing, co-localization studies, quantitative intensity analysis across a wide dynamic range. |
Table 2: Performance Characteristics in Drug Development Context
| Characteristic | Chromogenic Methods | Fluorescent Methods |
|---|---|---|
| Reproducibility (Inter-lab) | Moderate to High (standardized clinical protocols exist) | Variable to High (requires meticulous calibration) |
| Compatibility with Archived FFPE | Excellent (gold standard) | Good (subject to autofluorescence) |
| Throughput for Large Cohorts | High (automated scanners widespread) | Moderate (slower scanning, larger file sizes) |
| Objective Quantification Potential | High with advanced image analysis | Very High (direct intensity measurement) |
| Regulatory Acceptance | Very High (long history in companion diagnostics) | Increasing (especially in multiplexed assays) |
Detailed Experimental Protocols
Protocol 1: H-Score Quantification for Chromogenic DAB IHC
Purpose: To semi-quantitatively assess protein expression level and distribution in tissue sections.
- Image Acquisition: Scan stained slides at 20x magnification using a whole-slide brightfield scanner (e.g., Aperio AT2, Leica Aperio VERSA).
- Region of Interest (ROI) Annotation: Using analysis software (e.g., QuPath), annotate the relevant tissue compartments (tumor parenchyma, stroma).
- Color Deconvolution: Apply a color deconvolution algorithm (e.g., Ruifrok & Johnston method) to separate the DAB channel.
- Thresholding: Set an optical density threshold to distinguish "positive" from "negative" staining.
- Intensity Classification: Manually or algorithmically classify positive pixels into weak (1+), moderate (2+), and strong (3+) intensity bins based on OD.
- Calculation: For each ROI, calculate the H-Score: H-Score = Σ (Pi × i), where Pi is the percentage of cells stained at intensity i (1-3). The score ranges from 0 to 300.
Protocol 2: Mean Fluorescence Intensity (MFI) Quantification for Multiplex Fluorescent ICC
Purpose: To quantify the absolute expression level of multiple targets in cultured cells.
- Cell Seeding & Fixation: Seed cells on chambered coverslips. Fix with 4% PFA for 15 min and permeabilize with 0.1% Triton X-100.
- Multiplex Staining: Incubate with primary antibodies from different host species, followed by appropriate species-specific fluorophore-conjugated secondary antibodies (e.g., Alexa Fluor 488, 555, 647). Include a nuclear stain (DAPI or Hoechst).
- Image Acquisition: Acquire Z-stack images using a confocal or high-content fluorescence microscope (e.g., Zeiss LSM 980, PerkinElmer Opera Phenix). Use consistent exposure times and laser powers across all samples.
- Preprocessing: Apply flat-field correction and subtract background (based on no-primary-control slides).
- Segmentation: Use the nuclear stain to perform cell segmentation (identify individual cells). Expand the region to define cytoplasmic or membrane compartments.
- Intensity Measurement: For each cell and each fluorescence channel, measure the Mean Fluorescence Intensity (MFI) within the defined cellular compartment.
- Data Normalization: Normalize MFI values to an internal control (e.g., housekeeping protein) or a negative control population.
Signaling Pathway & Workflow Visualizations
Title: Chromogenic IHC Workflow for Quantification
Title: Multiplex Fluorescent ICC Workflow
Title: Quantitative Method Selection Logic Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Quantitative IHC/ICC
| Item | Function in Quantification |
|---|---|
| Validated Primary Antibodies | High specificity and lot-to-lot consistency are non-negotiable for reproducible quantitative data. |
| Polymer-based Detection Systems (e.g., HRP- or AP-Polymer) | Amplify signal with high sensitivity and low background, critical for both chromogenic and fluorescent tyramide signal amplification (TSA) methods. |
| Chromogen Kits (e.g., DAB, AEC) | Provide stable, precipitating substrates for brightfield detection. DAB is preferred for quantification due to permanence and OD properties. |
| Fluorophore-Conjugated Secondaries (e.g., Alexa Fluor series) | Offer bright, photostable signals across a range of wavelengths for multiplexing and intensity-based quantification. |
| Automated Slide Stainers (e.g., Leica BOND, Ventana Benchmark) | Standardize all staining steps (dewaxing, retrieval, incubation times) to minimize pre-analytical variability, the largest source of quantification error. |
| Whole-Slide Scanners (Brightfield & Fluorescent) | Digitize slides with consistent illumination and focus, creating high-resolution images suitable for pixel-level analysis. |
| Image Analysis Software (e.g., QuPath, HALO, Visiopharm, CellProfiler) | Perform color deconvolution, cell/tissue segmentation, and biomarker scoring based on user-defined algorithms. |
| Multispectral Imaging Systems (e.g., Akoya PhenoImager, RareCyte) | Acquire and unmix the full emission spectrum at each pixel, enabling high-plex fluorescence quantification on standard FFPE. |
| Certified Reference Materials (e.g., cell line microarrays, control tissue slides) | Serve as inter-assay and inter-laboratory calibration standards to ensure quantitative data is comparable over time and across sites. |
The "best" method for image quantification is contingent upon the primary research question framed within the chromogenic-fluorescent dichotomy. Chromogenic quantification (H-Score, positive pixel%) remains the robust, clinically-translatable choice for high-throughput, single-analyte studies where tissue morphology is paramount. Fluorescent quantification (MFI, co-localization) is superior for research requiring multiplex biomarker analysis, precise measurement across a broad dynamic range, or spatial biology applications. The emerging convergence—multiplex fluorescent staining coupled with multispectral imaging and subsequent conversion to brightfield-like images—promises to combine the strengths of both paradigms, offering a powerful path forward for next-generation quantitative biomarker discovery and scoring.
In the context of research comparing chromogenic (DAB) and fluorescent detection systems for immunohistochemistry (IHC) and immunocytochemistry (ICC), rigorous validation of sensitivity and specificity is paramount for both publication in peer-reviewed journals and for diagnostic assay development. This guide details the core principles and experimental protocols required to establish these key performance metrics.
Foundational Concepts: Sensitivity vs. Specificity in Detection
Sensitivity in IHC/ICC refers to the ability of the detection system to identify true-positive signals, minimizing false negatives. It is influenced by the signal amplification power and the signal-to-noise ratio. Specificity is the ability to correctly identify the absence of a target (true negatives), minimizing false-positive signals caused by non-specific binding or endogenous activity.
For chromogenic systems, signal is visualized as a colored precipitate, while fluorescent systems emit light at specific wavelengths upon excitation. The choice between them directly impacts these parameters.
Experimental Protocols for Validation
Protocol 2.1: Determining Analytical Sensitivity (Limit of Detection)
Objective: To establish the lowest target antigen concentration that can be reliably distinguished from background.
- Cell Line or Tissue Selection: Use a cell line with known, variable expression levels of the target antigen or a tissue microarray with graded expression.
- Serial Dilution: Create a series of sample preparations with antigen expression titrated via siRNA knockdown, competitive inhibition, or using a dilution series of a cell line pellet.
- Staining: Process all samples in the same run using the candidate detection system (chromogenic or fluorescent).
- Analysis (Chromogenic): Use digital image analysis to determine the optical density of stain per cell or area. The limit of detection (LoD) is the point where the mean signal is three standard deviations above the mean background of negative controls.
- Analysis (Fluorescent): Use fluorescence microscopy or scanners to measure mean fluorescence intensity (MFI). LoD is calculated similarly.
Protocol 2.2: Assessing Specificity (Background & Cross-Reactivity)
Objective: To quantify non-specific signal and confirm target specificity.
- Primary Antibody Controls:
- Negative Control: Omit primary antibody (use antibody diluent only).
- Isotype Control: Use an irrelevant immunoglobulin of the same species, class, and concentration as the primary antibody.
- Pre-absorption Control: Pre-incubate the primary antibody with a 10-fold molar excess of the target peptide/protein prior to staining.
- Detection System Controls:
- Endogenous Activity: For chromogenic IHC, include controls for endogenous peroxidase (with and without H₂O₂ blocking) and alkaline phosphatase.
- Endogenous Fluorescence: For fluorescent IHC/ICC, include unstained samples to assess autofluorescence.
- Secondary Antibody Only: Apply detection system without any primary antibody.
- Quantification: Measure background signal in control samples. Specificity is confirmed when signal in the test sample significantly exceeds all control sample signals.
Protocol 2.3: Direct Comparison of Chromogenic vs. Fluorescent Systems
Objective: To directly compare sensitivity and specificity profiles on identical samples.
- Sample Preparation: Use consecutive tissue sections or identical cell pellet arrays.
- Parallel Staining: Perform IHC/ICC for the same target antigen using optimized protocols for both a chromogenic (DAB) and a fluorescent (e.g., Tyramide Signal Amplification/TSA or standard fluorescent secondary) detection system.
- Multiplexing Consideration: For fluorescent systems, include a single-plex stain and a duplex stain with another marker to check for spectral bleed-through (specificity check).
- Quantitative Correlation: Use digital pathology platforms to quantify signal (Optical Density for DAB, MFI for fluorescent) from the same anatomical regions or cells. Perform linear regression analysis.
Data Presentation: Quantitative Comparison
Table 1: Performance Metrics of Chromogenic (DAB) vs. Fluorescent (TSA-Cy5) Detection for Target p53 in Model Cell Lines
| Metric | Chromogenic (DAB) System | Fluorescent (TSA-Cy5) System | Measurement Method |
|---|---|---|---|
| Limit of Detection (LoD) | 1:800 (Antibody Dilution Factor) | 1:3200 (Antibody Dilution Factor) | Serial AB dilution on low-expressing cell pellet |
| Signal-to-Background Ratio | 12.5 ± 2.1 | 45.3 ± 5.7 | Mean Signal (Positive) / Mean Signal (Isotype Control) |
| Dynamic Range | ~2 log units | ~4 log units | Linear range of optical density or MFI vs. antigen concentration |
| Multiplexing Capacity | 1 (with serial staining) | 4+ (simultaneous) | Number of distinct targets identifiable in one section |
| Spatial Resolution | Excellent, no signal diffusion | Excellent, but subject to optical limits | Visual assessment of subcellular localization |
| Background Source | Endogenous peroxidase, non-specific AB binding | Autofluorescence, non-specific AB binding, spectral bleed-through | - |
Table 2: Essential Research Reagent Solutions for Detection Validation
| Reagent / Solution | Function in Validation | Critical Consideration |
|---|---|---|
| Validated Primary Antibodies | Target specificity binder. | Use CRISPR-validated or pre-absorbed controls for specificity tests. |
| Isotype Control Antibodies | Distinguish specific from non-specific Fc receptor or protein binding. | Must match host species, immunoglobulin class, and concentration of primary. |
| Antigen Retrieval Buffers (pH 6 & 9) | Unmask epitopes formalin-fixed paraffin-embedded tissue. | pH optimization is target-specific and critical for both sensitivity and specificity. |
| Signal Amplification Systems (e.g., TSA) | Enhance sensitivity, especially for low-abundance targets. | Can increase background if not meticulously titrated; essential for fluorescent multiplexing. |
| Blocking Serums/Normal Serum | Reduce non-specific secondary antibody binding. | Should be from the same species as the secondary antibody host. |
| Enzyme Substrates (DAB, Vector NovaRED) | Produce insoluble chromogen precipitate for brightfield microscopy. | DAB is carcinogenic; use ready-to-use metal-enhanced substrates for higher sensitivity. |
| Fluorophore-Conjugated Secondaries (e.g., Alexa Fluor dyes) | Bind primary antibody and emit signal upon excitation. | Choose fluorophores matched to your microscope's filter sets and with minimal spectral overlap. |
| Nuclear Counterstains (Haematoxylin, DAPI, Hoechst) | Provide tissue/cellular architecture context. | DAPI/Hoechst must be compatible with fluorescent filter sets; haematoxylin for DAB. |
| Mounting Media (Aqueous, Antifade) | Preserve samples and optimize signal for imaging. | Use antifade (e.g., ProLong Diamond) for fluorescence to prevent photobleaching. |
| Autofluorescence Quenchers | Reduce non-specific background in fluorescent IHC. | Apply after staining but before mounting (e.g., Vector TrueVIEW, Sudan Black B). |
Visualization of Workflows and Pathways
Title: Comparative IHC/ICC Detection Workflow: Chromogenic vs Fluorescent
Title: Sensitivity & Specificity Relationship to Ground Truth
For diagnostic applications, chromogenic IHC (DAB) remains the standard due to its permanence, compatibility with standard histopathology workflows, and ease of interpretation. Validation requires stringent adherence to CLIA/CAP guidelines, emphasizing reproducibility and specificity.
For research publication, particularly in multiplex biomarker studies or spatial biology, fluorescent detection offers superior sensitivity, dynamic range, and multiplexing capability. Validation must include comprehensive controls for spectral cross-talk and autofluorescence.
Regardless of the system, a validation dossier for publication or diagnostics must present quantitative data on sensitivity (LoD) and specificity (background levels, control reactions) in a clear, tabular format, supported by representative images and a detailed methodology enabling replication.
This technical guide provides a detailed cost-benefit analysis (CBA) for immunohistochemistry (IHC) and immunocytochemistry (ICC) studies comparing chromogenic and fluorescent detection methods. Framed within a broader research thesis comparing these two fundamental detection paradigms, this document quantifies the investment in reagents, equipment, and time. The goal is to equip researchers and drug development professionals with data-driven decision-making tools for experimental design and resource allocation in preclinical and diagnostic research.
Foundational Concepts: Chromogenic vs. Fluorescent Detection
Chromogenic detection typically employs enzymes such as horseradish peroxidase (HRP) or alkaline phosphatase (AP) to catalyze the deposition of a colored precipitate (e.g., DAB, AEC, Fast Red) at the antigen site. Fluorescent detection relies on fluorophore-conjugated antibodies or probes that emit light of a specific wavelength upon excitation.
The choice between systems influences downstream costs, equipment needs, and workflow complexity. Chromogenic assays are generally viewed as lower-cost for single-plex, brightfield microscopy applications, while fluorescent assays offer superior multiplexing capabilities but require more sophisticated instrumentation and can suffer from photobleaching.
Quantitative Cost-Benefit Analysis
The following tables synthesize current market data and published protocol timings into a direct comparison. All monetary values are estimated in USD and represent list prices for research-scale quantities. Time investments are based on standard protocols for formalin-fixed, paraffin-embedded (FFPE) tissue sections.
Table 1: Reagent Cost Analysis per Sample (Single-Plex)
| Cost Component | Chromogenic (DAB) | Fluorescent (Direct, Alexa Fluor 488) | Notes |
|---|---|---|---|
| Primary Antibody | $2.50 - $10.00 | $2.50 - $10.00 | Largest variable; assumed identical for both. |
| Detection System | $3.00 - $5.00 | $0.50 - $2.00 | Cost for polymer-HRP/AP vs. fluorophore direct conjugate. |
| Chromogen/Substrate | $1.00 - $2.50 | N/A | DAB kit cost. |
| Counterstain & Mounting | $0.50 | $2.00 - $5.00 | Hematoxylin/Aqueous mount vs. DAPI/Antifade mount. |
| Total Estimated Cost | $7.00 - $18.00 | $5.00 - $17.00 | Direct fluorescence can be cheaper for single-plex. |
Table 2: Multiplexing (3-Plex) Cost & Time Comparison
| Parameter | Chromogenic Sequential | Fluorescent Simultaneous |
|---|---|---|
| Approx. Reagent Cost/Sample | $25 - $60 | $15 - $40 |
| Hands-on Time | 8 - 12 hours (over 2-3 days) | 4 - 6 hours (single day) |
| Total Protocol Duration | 2-3 days | 6-8 hours |
| Key Limitation | Antigen stripping required; signal overlap. | Spectral overlap requires panel optimization. |
Table 3: Capital Equipment & Operational Costs
| Equipment | Typical Use | Approx. Cost Range | Necessity |
|---|---|---|---|
| Brightfield Microscope | Chromogenic imaging | $10k - $50k | Essential for chromogenic. |
| Standard Epifluorescence Microscope | Fluorescent imaging | $30k - $80k | Essential for fluorescent. |
| Confocal Microscope | High-res fluorescent imaging | $150k - $500k | Needed for advanced multiplexing. |
| Slide Scanner | Digital pathology, quantitation | $80k - $250k | Beneficial for both, higher throughput. |
| Specialized Software (Quantitation) | Image analysis | $5k - $20k (licenses) | Higher need/complexity for fluorescent multiplex. |
Detailed Experimental Protocols
Protocol 1: Sequential Chromogenic Multiplex IHC (FFPE)
Principle: Sequential application of primary antibodies, enzyme-labeled polymers, and chromogens, with heat-induced epitope retrieval (HIER) stripping steps between each round.
- Deparaffinize, rehydrate, and perform HIER (e.g., citrate buffer, pH 6.0).
- Apply Protein Block (10% serum, 10 min).
- Apply Primary Antibody #1 (species A, 1 hr, RT).
- Apply HRP-Polymer conjugate vs. species A (30 min, RT).
- Develop with DAB chromogen (5-10 min).
- Antigen Stripping: Heat slides in HIER buffer (95-100°C, 20 min).
- Repeat steps 2-5 for Primary Antibody #2 (species B) with a different chromogen (e.g., Fast Red).
- Counterstain with Hematoxylin, aqueous mount.
Protocol 2: Simultaneous Fluorescent Multiplex IHC (FFPE)
Principle: Concurrent application of primary antibodies raised in different hosts, detected with species-specific fluorophore-conjugated secondary antibodies.
- Deparaffinize, rehydrate, perform HIER.
- Apply Protein Block (e.g., 3% BSA, 30 min).
- Apply Primary Antibody Cocktail (mouse anti-Protein X, rabbit anti-Protein Y; 1-2 hr, RT or overnight at 4°C).
- Apply Secondary Antibody Cocktail (e.g., AF488-conjugated donkey anti-mouse, AF555-conjugated donkey anti-rabbit; 1 hr, RT, in darkness).
- Apply DAPI nuclear stain (5 min).
- Mount with ProLong Antifade mounting medium, cure overnight.
Signaling Pathways & Workflow Diagrams
Diagram Title: Chromogenic Detection Signaling Pathway
Diagram Title: Fluorescent Detection Signaling Pathway
Diagram Title: Detection Method Selection Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in IHC/ICC | Key Consideration for CBA |
|---|---|---|
| Polymer-Based Detection Systems (e.g., HRP-Polymer) | Amplifies signal by conjugating multiple enzyme molecules to a secondary antibody backbone. Higher sensitivity than direct conjugates. | Chromogenic cornerstone. Increases per-test cost but reduces antibody consumption and improves consistency. |
| Tyramide Signal Amplification (TSA) Reagents | Enzyme-catalyzed deposition of fluorophore-tyramide for massive signal amplification. Enables high-plex fluorescence. | Significant reagent cost increase, but enables use of low-abundance antibodies and ultra-plexing (10+ targets). |
| Multiplex-Optimized Antibody Panels | Pre-validated primary antibody cocktails from distinct host species. | High upfront cost and vendor lock-in, but saves months of validation time and reduces risk of cross-reactivity. |
| Automated Staining Platforms | Instrumentation for consistent, hands-off antibody and reagent application. | High capital cost (>$50k), but drastically improves reproducibility, reduces labor time, and standardizes protocols for high-throughput studies. |
| Antigen Retrieval Buffers (Citrate, EDTA, Tris-EDTA) | Unmask epitopes cross-linked by fixation. Critical for FFPE specimens. | Low cost. Choice of buffer pH significantly impacts outcome and must be optimized per target; failure wastes all downstream reagents. |
| Antifade Mounting Media (e.g., ProLong, Vectashield) | Preserves fluorescence by reducing photobleaching. Contains DAPI for nuclear counterstain. | Essential for fluorescence. Cost is 5-10x higher than aqueous mounting media for chromogenic slides. |
This whitepaper examines the evolution of multiplexed imaging technologies within the established context of chromogenic versus fluorescent detection in immunohistochemistry (IHC) and immunocytochemistry (ICC). While chromogenic IHC (single-plex, enzyme-based colorimetric detection) has been the diagnostic mainstay, and standard fluorescent IHC/ICC enabled early multiplexing, emerging platforms now allow for the simultaneous, high-parameter visualization of dozens of biomarkers within intact tissue architecture. This shift is critical for advancing research in immunology, oncology, and complex disease pathology, providing unparalleled spatial phenotyping for drug development.
Core Technology Comparison: Chromogenic, Fluorescent, and Hyperplex Platforms
Table 1: Quantitative Comparison of IHC/ICC Detection Modalities
| Feature | Chromogenic IHC/ICC | Standard Fluorescent IHC/ICC | Hyperplex Imaging (e.g., CODEX, MIBI) |
|---|---|---|---|
| Maxplex (Theoretical) | 1-2 (sequential) | 4-8 (simultaneous) | 40+ (CODEX), 50+ (MIBI) |
| Spatial Context | Preserved | Preserved | Preserved with subcellular detail |
| Detection Method | Enzyme (HRP/AP) → Chromogen | Fluorophore Direct/Indirect Excitation | Oligo-labeled Abs → Cyclic Imaging |
| Signal Background | High (endogenous pigments) | Medium (autofluorescence) | Low (removal via washing) |
| Quantification | Semi-quantitative (density) | Quantitative (intensity) | Highly Quantitative (counts/intensity) |
| Throughput | High | Medium | Low to Medium (acquisition/processing) |
| Primary Application | Diagnostic pathology, single targets | Research, limited multiplex | High-plex spatial phenotyping, systems biology |
Detailed Methodologies for Key Hyperplex Experiments
Protocol 1: CODEX (CO-Detection by indEXing) Staining and Data Acquisition
Principle: Antibodies are conjugated to unique oligonucleotide barcodes. Sequential fluorescence in situ hybridization of complementary, dye-labeled reporters enables cyclic imaging.
Procedure:
- Tissue Preparation: Formalin-fixed, paraffin-embedded (FFPE) tissue sections (4-5 µm) are mounted and baked. Standard deparaffinization and antigen retrieval are performed.
- Antibody Staining: A cocktail of ~50+ oligonucleotide-conjugated primary antibodies is applied to the sample overnight at 4°C.
- Instrument Setup: Load sample into the CODEX instrument (fluidics system integrated with an automated microscope).
- Cyclic Imaging:
- A first set of fluorescent reporters (e.g., 3 dyes) complementary to a subset of barcodes is introduced.
- The entire region of interest (ROI) is imaged (DAPI, FITC, Cy3, Cy5 channels).
- A gentle chemical wash cleaves and removes the fluorophores without disturbing antibody binding.
- The next set of reporters is introduced, and the cycle repeats (typically 15+ cycles).
- Image Processing & Deconvolution: Computational stitching and deconvolution align cycles and generate a single, high-plex image where each pixel contains a 50+ dimensional vector of biomarker expression.
Protocol 2: Multiplexed Ion Beam Imaging (MIBI) Staining and Analysis
Principle: Antibodies are labeled with rare-earth metal isotopes, not fluorophores. The tissue is ablated by a primary ion beam, and the ejected secondary ions (the metal tags) are quantified by time-of-flight mass spectrometry.
Procedure:
- Tissue Preparation & Staining: FFPE tissue is prepared as above. Staining is performed with a cocktail of antibodies conjugated to pure elemental lanthanide isotopes (e.g., Nd150, Dy161) via polymer cheators.
- Vacuum Chamber Placement: The stained sample is placed in the high-vacuum chamber of the MIBI-TOF instrument.
- Raster Scanning & Ablation: A focused primary oxygen ion beam raster-scans the tissue pixel by pixel. At each pixel, the beam ablates the tissue, releasing the metal tags.
- Mass Spectrometry Detection: The ejected secondary ions are filtered and quantified by a time-of-flight mass spectrometer. Each isotope's mass identifies its corresponding antibody.
- Data Reconstruction: The mass spectrometry data for each pixel is reconstructed into a quantitative, multi-channel image. Signal is absolute ion counts, not photon intensity.
Visualizing Workflows and Pathways
CODEX Experimental Workflow
MIBI-TOF Imaging Principle
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Hyperplex Imaging Experiments
| Item | Function in Experiment | Example/Notes |
|---|---|---|
| Oligonucleotide-Conjugated Antibodies (CODEX) | Primary detection reagent; each antibody has a unique DNA barcode. | Commercially available panels or custom conjugation kits. |
| Fluorophore-Labeled Reporters (CODEX) | Complementary oligonucleotides with fluorescent dyes for cyclic detection. | Typically 3-4 fluorophores (e.g., FITC, Cy3, Cy5) cycled over many rounds. |
| Metal-Tagged Antibodies (MIBI/IMC) | Primary detection reagent; antibodies conjugated to pure elemental isotopes. | Lanthanide isotopes (e.g., 141Pr, 153Eu, 165Ho) via MAXPAR polymer. |
| CODEX Fluidics Instrument | Automated system for cyclic reagent addition, washing, and imaging. | Akoya Biosciences CODEX system. |
| MIBI-TOF or Imaging Mass Cytometer | Instrument for ablation and detection of metal isotopes from tissue. | Standard IMC (Fluidigm/Hyperion) or MIBIscope. |
| High-Plex Image Analysis Software | For image registration, single-cell segmentation, and phenotyping. | Akoya Phenoptics, Visiopharm, HALO, or open-source (CellProfiler, QuPath). |
| Antigen Retrieval Buffer (pH 6 or 9) | Unmasks epitopes in FFPE tissue for antibody binding. | Critical for successful high-plex staining. |
| Validated Antibody Panel | Pre-tested antibody cocktail ensuring specificity and lack of cross-reactivity. | Requires extensive optimization for concentration and pairwise interactions. |
Conclusion
Selecting between chromogenic and fluorescent detection is not a one-size-fits-all decision but a strategic choice dictated by the research question, required output, and available resources. Chromogenic IHC remains the gold standard for clinical pathology due to its permanence and compatibility with brightfield microscopy, while fluorescent methods are indispensable for multiplexing, co-localization, and quantitative analysis. Successful implementation requires a deep understanding of each method's principles, meticulous optimization, and rigorous validation. As biomedical research advances toward highly multiplexed spatial biology and quantitative digital pathology, fluorescence-based and next-generation multiplexing platforms are gaining prominence. However, the simplicity and diagnostic clarity of chromogenic stains ensure their enduring role. Researchers must stay informed on these evolving technologies to leverage the full potential of IHC and ICC in driving discovery and translational medicine.