Direct, Indirect, and Sandwich ELISA: A Comprehensive Comparison for Researchers
This article provides a detailed comparative analysis of three core ELISA formats—Direct, Indirect, and Sandwich—tailored for researchers, scientists, and drug development professionals.
Direct, Indirect, and Sandwich ELISA: A Comprehensive Comparison for Researchers
Abstract
This article provides a detailed comparative analysis of three core ELISA formats—Direct, Indirect, and Sandwich—tailored for researchers, scientists, and drug development professionals. It covers foundational principles, methodological steps, and specific applications for each variant. The scope extends to practical guidance on troubleshooting common issues, optimizing assay performance, and validating results through a critical comparison of sensitivity, specificity, and cost-effectiveness. The objective is to serve as a definitive guide for selecting and implementing the most appropriate ELISA methodology for diverse research and diagnostic goals.
ELISA Fundamentals: Principles, History, and Core Concepts
What is an ELISA? Defining the Enzyme-Linked Immunosorbent Assay
Enzyme-Linked Immunosorbent Assay (ELISA) is a fundamental immunochemical biochemical assay used to detect and quantify substances such as peptides, proteins, antibodies, and hormones in complex biological samples [1] [2]. The method is based on the principle of detecting antigen-antibody interactions, where an enzyme-linked conjugate reacts with a substrate to produce a measurable signal, typically a color change [3] [2]. First developed in the 1970s, ELISA has become a cornerstone technique in research and diagnostic laboratories worldwide due to its high sensitivity and specificity [2].
This guide provides an objective comparison of the four main ELISA variants—direct, indirect, sandwich, and competitive—to help researchers select the optimal method for their specific applications.
Core Principles and Components of an ELISA
At its core, ELISA depends on the highly specific binding between an antibody and its target antigen. The assay is typically performed on a solid phase, such as a 96-well polystyrene microplate, to which antigens or antibodies are immobilized [1] [2]. This solid-phase design facilitates the separation of bound and unbound materials through simple washing steps, making ELISA robust even for crude sample preparations [1].
The key components essential for any ELISA protocol include [2]:
- Solid Phase: Usually a 96-well microplate, which provides a surface for immobilizing the target molecule.
- Capture Molecule: Either an antigen or an antibody that is immobilized to the solid phase to "capture" the analyte of interest from the sample.
- Detection Antibody: An antibody that binds specifically to the captured analyte. This antibody is often linked (conjugated) to an enzyme such as Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP).
- Substrate: A chemical compound that is converted by the enzyme into a detectable product, leading to a colorimetric, fluorescent, or chemiluminescent signal.
- Signal Measurement: The intensity of the final signal is measured using a plate reader and is proportional to the amount of analyte in the sample.
The following diagram illustrates the general logical relationship and workflow common to all ELISA types, from sample immobilization to signal detection.
Comparative Analysis of Major ELISA Types
ELISA methods are primarily categorized into four types, each with distinct mechanisms, advantages, and limitations. The table below provides a structured, quantitative comparison to guide your selection.
| ELISA Type | Detection Strategy | Sensitivity | Specificity | Steps Required | Best For / Optimal Use Cases |
|---|---|---|---|---|---|
| Direct ELISA [4] [5] | Enzyme-labeled primary antibody binds directly to the immobilized antigen. | Low (no signal amplification) [5]. | High (avoids cross-reactivity from secondary antibodies) [4]. | Fewest (fewer steps, faster protocol) [4] [5]. | Analyzing antigen-antibody immunoreactivity [5]. |
| Indirect ELISA [4] [5] | Unlabeled primary antibody binds antigen, then is detected by an enzyme-labeled secondary antibody. | High (signal amplification via multiple secondary antibodies binding to a single primary) [4] [5]. | Moderate (potential for cross-reactivity from secondary antibody) [4] [5]. | More than Direct (additional incubation and wash step) [5]. | Detecting and quantifying total antibody levels in serum [3] [5]. |
| Sandwich ELISA [6] [4] | Antigen is "sandwiched" between a capture antibody and a detection antibody. | Very High (2–5 times more sensitive than direct/indirect) [5]. | Very High (requires two specific antibodies for a single antigen) [4] [5]. | Most (requires careful optimization of matched antibody pairs) [4] [5]. | Measuring complex or impure samples without antigen purification (e.g., cytokines in cell culture media) [4] [5]. |
| Competitive ELISA [4] [5] | Sample antigen and labeled reference antigen compete for a limited number of antibody-binding sites. | Moderate (overall sensitivity and specificity are lower) [4]. | Moderate | Highly flexible (can be based on direct, indirect, or sandwich formats, but often complex) [5]. | Detecting small antigens or when only one specific antibody is available [3] [5]. |
The following diagram visualizes the fundamental differences in the antigen-antibody binding strategies for each of the four main ELISA types.
Detailed Experimental Protocols
To ensure reproducible and high-quality results, adherence to standardized protocols is critical. Below are the detailed step-by-step methodologies for the most common ELISA formats.
Sandwich ELISA Protocol
The sandwich ELISA is renowned for its high sensitivity and specificity and is widely considered the gold standard for quantitative protein detection [3]. The following workflow details its key steps.
Step-by-Step Procedure [6] [3] [1]:
- Coating: Dilute the capture antibody in a coating buffer (e.g., carbonate-bicarbonate buffer, pH 9.4 or phosphate-buffered saline [PBS], pH 7.4) to a concentration typically between 2-10 μg/mL. Add the solution to a polystyrene microplate (≥ 100 μL/well for a 96-well plate). Incubate for several hours at 37°C or overnight at 4°C.
- Blocking: Discard the coating solution. Add a blocking buffer (e.g., 1-5% BSA, non-fat dry milk, or casein in PBS) to cover all unsaturated binding sites on the plastic surface. Incubate for 1-2 hours at room temperature. Wash the plate 2-3 times with a wash buffer (e.g., PBS or Tris-buffered saline containing a mild detergent like 0.05% Tween 20).
- Sample Incubation: Add the sample or antigen standard, diluted in blocking or sample buffer, to the wells. Incubate for at least 1-2 hours at room temperature to allow the antigen to be captured by the immobilized antibody. Wash as in Step 2 to remove unbound antigens and other sample components.
- Detection Antibody Incubation: Add the enzyme-conjugated detection antibody, specific to a different epitope on the antigen, diluted in blocking buffer. Incubate for 1-2 hours at room temperature. Wash thoroughly as before to remove any unbound detection antibody.
- Signal Development: Add the enzyme substrate solution (e.g., TMB for HRP). Incubate in the dark for 15-30 minutes, allowing color development.
- Signal Measurement: Stop the enzyme-substrate reaction by adding an acidic stop solution (e.g., 1M H₂SO₄ for TMB, which changes the color from blue to yellow). Immediately measure the absorbance (Optical Density, OD) of each well using a plate reader at the appropriate wavelength (e.g., 450 nm for TMB).
Competitive ELISA Protocol
Competitive ELISA is particularly useful for detecting small molecules or when only one specific antibody is available [3] [5].
Step-by-Step Procedure [4] [1]:
- Coating: Coat the microplate with a known amount of purified antigen (or the specific antibody, depending on the format). Incubate and block as described in the sandwich ELISA protocol.
- Competition Reaction: Pre-incubate a constant amount of the enzyme-labeled antibody (or labeled antigen) with the sample containing the unknown antigen. Alternatively, add both the labeled reagent and the sample simultaneously to the coated plate. During this incubation, the antigen in the sample (unlabeled) and the immobilized antigen (or the labeled antigen) compete for binding to the limited number of antibody-binding sites.
- Washing: Wash the plate to remove any unbound components, including the unbound labeled antigen-antibody complexes.
- Signal Development and Measurement: Add substrate and measure the signal as described previously. The key differentiator is that the signal intensity is inversely proportional to the concentration of the antigen in the sample. A higher antigen concentration in the sample leads to less labeled reagent being bound and, consequently, a weaker signal.
The Scientist's Toolkit: Essential Research Reagent Solutions
Establishing a reliable ELISA requires a suite of high-quality reagents and equipment. The following table details the essential materials and their functions.
| Item Category | Specific Examples | Critical Function in the Assay |
|---|---|---|
| Solid Phase [1] [2] | 96- or 384-well polystyrene microplates | Provides the solid surface for immobilizing the capture molecule (antigen or antibody). |
| Capture & Detection Molecules [3] [1] | Monoclonal/polyclonal antibodies, purified antigens | Key reagents that provide the assay's specificity by binding to the target analyte. |
| Enzyme Conjugates [1] [2] | Horseradish Peroxidase (HRP), Alkaline Phosphatase (AP) | Enzymes linked to antibodies; catalyze the conversion of substrate to a detectable signal. |
| Substrates [1] [2] | TMB (3,3',5,5'-Tetramethylbenzidine), pNPP (para-Nitrophenyl Phosphate) | Chemicals converted by enzyme conjugates to produce a colorimetric, fluorescent, or chemiluminescent signal. |
| Buffers [1] | Coating buffer (e.g., carbonate, PBS), Blocking buffer (e.g., BSA, casein), Wash buffer (e.g., PBS-Tween) | Enable proper immobilization, reduce non-specific background, and remove unbound material. |
| Laboratory Equipment [6] [2] | ELISA plate reader (spectrophotometer), plate washer, micropipettes | Precisely dispense reagents, automate washing steps, and accurately quantify the output signal. |
The selection of an appropriate ELISA format is a critical determinant of experimental success. The direct ELISA offers simplicity and speed, while the indirect format provides enhanced sensitivity and flexibility. The sandwich ELISA remains the gold standard for specificity and sensitivity in quantifying proteins from complex mixtures, and the competitive ELISA is the method of choice for small molecules or limited reagent availability.
By understanding the comparative strengths, weaknesses, and specific protocols of each method, researchers and drug development professionals can make an informed choice, ensuring robust, reliable, and meaningful data generation in both basic research and clinical diagnostics.
The Radioimmunoassay Revolution and the Emergence of ELISA
The development of radioimmunoassay (RIA) in the 1950s represented a breakthrough in biochemical measurement, enabling scientists to quantify hormones, drugs, and other biologically active substances with unprecedented sensitivity. This method utilized radioactive isotopes as labels to detect antigen-antibody interactions, allowing for the detection of extremely low concentrations of analytes in complex biological fluids [7]. For decades, RIA remained the gold standard for sensitive detection in clinical and research settings.
However, growing concerns about the health risks and regulatory burdens associated with handling radioactive materials created demand for safer alternatives [8]. This need catalyzed the development of enzyme-linked immunosorbent assay (ELISA) in the 1960s and early 1970s, pioneered by researchers including Engvall, Perlmann, Van Weemen, and Schuurs [2] [9]. The fundamental innovation was replacing radioactive labels with enzyme conjugates that could generate measurable color changes when exposed to appropriate substrates [9]. This transition from radioactivity to enzymatic detection established a new paradigm in immunoassays—one that maintained high sensitivity while eliminating radiation hazards.
A pivotal 1983 study directly compared both methods for detecting antibodies to chromatin components, finding that RIA could detect a human test antiserum at dilutions up to 1:102,400, compared to only 1:3,200 for ELISA, demonstrating RIA's superior sensitivity at the time but also highlighting ELISA's potential as a viable, safer alternative [10]. The first ELISA methodologies employed chromogenic reporters, but the technique has since evolved to incorporate fluorogenic, quantitative PCR, and electrochemiluminescent reporters [9]. The most recent developments include nanoparticle-based reporters that generate color signals visible to the naked eye [9].
Comparative Analysis of Immunoassay Techniques
The transition from RIA to ELISA represented a significant advancement, though each method maintains distinct advantages depending on application requirements.
Table 1: Comparison of Key Immunoassay Platforms
| Assay Type | Detection Principle | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|---|
| Radioimmunoassay (RIA) | Radioactive isotopes | Very High [10] [7] | High sensitivity for low analyte levels [7] | Radioactive hazards; specialized handling/equipment [7] |
| Enzyme-Linked Immunosorbent Assay (ELISA) | Enzyme-colorimetric reaction | High [11] [9] | Cost-effective; safer; high-throughput capability [7] | Generally lower sensitivity than RIA [10] |
| Chemiluminescent Immunoassay (CLIA) | Enzyme-chemical luminescence | Very High [7] | Broad dynamic range; automation friendly [7] | Higher cost; specialized equipment [7] |
| Fluoroimmunoassay (FIA) | Fluorescent compounds | Variable [7] | Fast analysis; highly sensitive with specific probes [7] | Sample matrix interference; limited dynamic range [7] |
While RIA demonstrated superior sensitivity in early comparisons, modern ELISA technologies have narrowed this gap through various amplification systems while offering superior safety profiles and operational convenience. A critical advantage of ELISA over Western blot is its completely quantitative results compared to the semi-quantitative nature of Western blots [11].
Modern ELISA Formats: Principles, Protocols, and Performance Characteristics
The evolution of ELISA technology has produced four principal formats, each with distinct mechanisms and applications suited to different experimental needs.
Direct ELISA
The simplest ELISA format involves immobilizing the antigen directly onto the microplate surface, followed by incubation with an enzyme-conjugated primary antibody that binds specifically to the target antigen [11] [9]. After washing to remove unbound antibody, substrate is added to produce a measurable color change.
Key Protocol Steps:
- Coat microplate with antigen sample (1-2 hours at 37°C or overnight at 4°C)
- Block remaining binding sites with BSA or other proteins (1-2 hours at room temperature)
- Add enzyme-conjugated primary antibody (1-2 hours at room temperature)
- Wash plate to remove unbound antibody
- Add enzyme substrate and measure color development [9] [12]
Advantages: Rapid protocol with fewer steps; eliminates secondary antibody cross-reactivity [11] [12] Disadvantages: Potentially lower sensitivity; higher background from sample proteins; requires labeled primary antibodies for each target [11] [9]
Indirect ELISA
This format introduces an amplification step to increase sensitivity. The antigen is immobilized on the plate, followed by incubation with an unlabeled primary antibody. A secondary antibody conjugated with an enzyme is then added, which binds to the primary antibody [11] [9].
Key Protocol Steps:
- Coat microplate with antigen
- Block nonspecific sites
- Add specific primary antibody
- Add enzyme-conjugated secondary antibody
- Wash and add substrate for detection [9]
Advantages: Enhanced sensitivity through signal amplification; versatile—same labeled secondary can detect various primary antibodies; preserves immunoreactivity of primary antibody [11] [12] Disadvantages: Potential for cross-reactivity with secondary antibody; additional incubation step required [11] [12]
Sandwich ELISA
As the most sensitive ELISA format, this technique captures the target antigen between two specific antibodies—a capture antibody immobilized on the plate and a detection antibody that completes the "sandwich" [11] [9]. This format requires carefully matched antibody pairs that recognize different epitopes on the target antigen.
Key Protocol Steps:
- Coat plate with capture antibody (overnight at 4°C)
- Block plate with BSA (1-2 hours at room temperature)
- Add sample containing target antigen (90 minutes at 37°C)
- Add primary detection antibody (1-2 hours at room temperature)
- Add enzyme-conjugated secondary antibody (1-2 hours at room temperature)
- Wash and add substrate [9]
Advantages: Highest sensitivity and specificity; compatible with complex samples [11] [12] Disadvantages: Time-consuming protocol; requires matched antibody pairs [11] [9]
Competitive ELISA
This format is particularly useful for detecting small molecules with single epitopes. The sample antigen and labeled reference antigen compete for binding to a limited amount of capture antibody. The signal produced is inversely proportional to the amount of antigen in the sample [11] [12].
Advantages: Effective for small molecules; requires less sample purification [11] [12] Disadvantages: Lower specificity; requires conjugated antigen [11]
Experimental Comparisons: Performance Variations Across Commercial ELISA Kits
Recent comparative studies highlight significant performance differences between commercial ELISA kits, emphasizing the importance of careful kit selection and validation.
Corticosterone Measurement in Serum Samples
A 2017 study compared four commercial ELISA kits for quantifying corticosterone in rat serum [8]. The same serum samples yielded significantly different values across kits, with the Arbor Assays kit measuring 357.75 ± 210.52 ng/mL compared to 40.25 ± 39.81 ng/mL for the DRG-5186 kit [8]. Despite high correlations between kits, the absolute concentration values varied substantially, indicating that relative differences within studies may be more reliable than absolute values across different kits [8].
Detection of Bordetella pertussis Antibodies
A recent comparison of six commercial ELISA kits and one in-house assay for detecting anti-pertussis antibodies revealed marked variations in sensitivity and specificity [13]. When testing 40 serum samples, IgA antibodies were detected in 27.5% of samples with the Savyon kit, 25.0% with Euroimmun, 20.0% with the in-house assay, but only 5.0% with the TestLine kit [13]. The study attributed these discrepancies to differences in antigen purity, calibration standards, and cutoff values between manufacturers [13].
SARS-CoV-2 Antibody Detection in Animal Sera
A 2025 evaluation of three ELISA kits for detecting SARS-CoV-2 antibodies in multiple animal species demonstrated that kits targeting the receptor binding domain (RBD) outperformed those detecting nucleoprotein antibodies [14]. ELISA-1 (RBD-targeting) showed the highest diagnostic performance, while ELISA-3 (nucleoprotein-targeting) demonstrated lower sensitivity for detecting seropositive animals [14]. This highlights how antigen choice significantly impacts assay performance, especially across species.
Table 2: Essential Research Reagent Solutions for ELISA
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Solid Phase | 96-well polystyrene microplates [2] [12] | Protein binding capacity >400ng/cm²; low CV (<5%) critical for reproducibility [12] |
| Coating Buffers | Carbonate-bicarbonate (pH 9.4), PBS (pH 7.4) [12] | Optimal pH conditions for antigen/antibody immobilization |
| Blocking Agents | BSA, ovalbumin, aprotinin, animal sera [9] | Cover unsaturated binding sites to minimize nonspecific background |
| Detection Enzymes | Horseradish peroxidase (HRP), Alkaline phosphatase (AP) [2] [9] | Generate measurable signals; HRP most common with multiple substrate options |
| Enzyme Substrates | TMB (colorimetric), CDP-Star (chemiluminescent) [9] [12] | Convert enzyme activity to detectable signals; choice affects sensitivity |
| Stop Solutions | HCl, H₂SO₄, NaOH [2] | Halt enzyme-substrate reaction at optimal timepoint |
The evolution from RIA to modern ELISA represents a paradigm shift in immunoassay technology, balancing the imperative for sensitive detection with practical considerations of safety, cost, and throughput. While RIA maintains advantages for certain applications requiring ultra-sensitive detection, ELISA has become the workhorse of modern diagnostics and research due to its versatility, safety profile, and continuous technical improvements.
Current trends suggest ELISA technology will continue evolving toward increased automation, integration with digital platforms, and enhanced sensitivity through novel signal amplification techniques [15]. The 2025 marketplace features diverse providers specializing in different applications, from high-throughput clinical screening to specialized research applications [15]. Future developments will likely focus on multiplexing capabilities and miniaturization to meet growing demands for comprehensive biomarker profiling from limited sample volumes [15].
For researchers and drug development professionals, selection of ELISA formats and commercial kits requires careful consideration of target analyte characteristics, required sensitivity, sample matrix effects, and intended application. The substantial variability between commercial kits underscores the necessity of thorough validation using appropriate standards and controls to ensure data reliability and reproducibility across laboratories.
The Enzyme-Linked Immunosorbent Assay (ELISA) is a foundational technique in biomedical research and clinical diagnostics, enabling the detection and quantification of soluble targets like peptides, proteins, antibodies, and hormones. [12] Its operation depends on the precise interplay of four core components: the solid-phase plate, antibodies, enzymes, and substrates. [2] The solid phase, typically a 96-well microplate, serves as the platform for immobilizing the assay reactants. [2] [9] Antibodies provide the critical specificity required to identify the target molecule from a complex mixture. [12] Finally, enzyme and substrate pairs generate a measurable signal, such as a color change, that is proportional to the amount of target present. [2] [9] The quality and compatibility of these components directly determine the performance of an ELISA, impacting its sensitivity, specificity, and reproducibility. This guide provides a detailed comparison of these core components, offering experimental data and protocols to inform their selection for various research applications, particularly within the context of comparing direct, indirect, and sandwich ELISA formats.
Comparative Analysis of Core Components
Solid-Phase Plates
The solid-phase plate is the physical foundation of the ELISA, providing a surface for the immobilization of antigens or antibodies. The choice of plate material and its surface properties are crucial for maximizing the assay's binding capacity and consistency.
- Materials and Binding: The most common materials are polystyrene, polyvinyl, and polypropylene. [2] Proteins bind to these plastics via hydrophobic interactions. [12] Polystyrene is the most frequently used due to its excellent optical clarity for colorimetric detection and high protein-binding capacity, typically exceeding 400 ng/cm². [12]
- Plate Selection: Clear polystyrene plates are standard for colorimetric detection. For fluorescent or chemiluminescent signals, black or white opaque plates are used to minimize background interference or to enhance light signal capture, respectively. [12]
- Performance Metrics: A key specification is the coefficient of variation (CV) for protein binding, which should be low (<5% is preferred) to ensure well-to-well and plate-to-plate reproducibility. [12] Imperfections or scratches in the plastic can cause significant aberrations in data acquisition. [12]
Table 1: Comparison of Solid-Phase Plate Properties
| Property | Polystyrene | Polyvinyl | Polypropylene |
|---|---|---|---|
| Common Use | Standard colorimetric ELISA | Similar to polystyrene | Specialized applications |
| Optical Clarity | High (clear) | Variable | Often opaque |
| Protein Binding Capacity | High (>400 ng/cm²) [12] | High | High |
| Typical CV | <5% (preferred) [12] | Information Missing | Information Missing |
| Best For | Colorimetric detection; general use | General use | Fluorescent/Chemiluminescent detection |
Antibodies
Antibodies are the targeting molecules of ELISA, defining its specificity. Their selection and configuration vary significantly between the main ELISA formats.
- Direct vs. Indirect Detection: In a direct ELISA, a single enzyme-conjugated primary antibody is used, making the protocol faster and eliminating potential cross-reactivity from a secondary antibody. [11] [12] However, it is less sensitive, and conjugating primary antibodies is time-consuming and expensive. [12] [16] In an indirect ELISA, an unlabeled primary antibody binds the antigen, and is then detected by an enzyme-conjugated secondary antibody. This allows for signal amplification (as multiple secondaries can bind a single primary) and greater flexibility, as the same labeled secondary can be used with various primary antibodies. [12] [17] The trade-off is an increased risk of cross-reactivity and a longer protocol. [11] [16]
- Sandwich ELISA Pairs: The sandwich ELISA requires two antibodies that bind to distinct, non-overlapping epitopes on the target antigen. [18] One serves as the capture antibody (immobilized on the plate) and the other as the detection antibody. [11] [18] This format provides the highest specificity and sensitivity and is ideal for complex samples, as non-target material can be washed away after the capture step. [11] [12] [17] The main challenge is the need for a "matched pair" of antibodies that do not interfere with each other's binding. [9] [12]
- Antibody Types: Monoclonal antibodies offer high specificity to a single epitope, making them ideal for sandwich assays. [18] Polyclonal antibodies, which recognize multiple epitopes, can increase sensitivity but may have higher cross-reactivity. [18] Recombinant antibodies are increasingly favored for their superior reproducibility and minimal batch-to-batch variation. [18]
Table 2: Antibody Roles in Different ELISA Formats
| ELISA Format | Primary Antibody | Secondary Antibody | Key Characteristic |
|---|---|---|---|
| Direct | Enzyme-conjugated | Not used | Single, labeled antibody; simple but less sensitive. [11] [12] |
| Indirect | Unlabeled | Enzyme-conjugated | Signal amplification; flexible but potential for cross-reactivity. [12] [17] |
| Sandwich | Capture antibody (unlabeled) | Detection antibody (may be conjugated or detected by a tertiary conjugate) | Two antibodies for high specificity; requires matched pairs. [9] [18] |
Enzymes and Substrates
The enzyme-substrate system is the signaling engine of the ELISA, converting the antibody-antigen binding event into a quantifiable signal.
- Common Enzymes: The two most widely used enzymes are Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP). [9] [12] HRP is smaller (44 kDa) and has a faster catalytic rate, while AP is known for its high stability. [2] [9] Other enzymes like β-galactosidase are used less frequently. [12]
- Substrates and Detection: The choice of substrate depends on the required sensitivity and the detection instrument available (spectrophotometer, fluorometer, or luminometer). [12]
- Colorimetric: For HRP, TMB (3,3',5,5'-Tetramethylbenzidine) is a common chromogenic substrate that produces a blue color, which turns yellow when stopped with a strong acid. [2] [9] Its absorbance is read at 450 nm. [2] For AP, pNPP (p-Nitrophenyl Phosphate) is a common substrate that produces a yellow product, measurable at 405 nm. [9]
- Other Types: Chemiluminescent substrates (which produce light) offer higher sensitivity than colorimetric ones, while fluorescent substrates enable detection with fluorometers. [17]
- Stop Solution: The enzyme-substrate reaction is typically terminated after 30-60 minutes using an acidic (e.g., H₂SO₄ or HCl) or basic (e.g., NaOH) solution. [2] This stabilizes the signal for measurement.
Table 3: Common Enzyme-Substrate Systems in ELISA
| Enzyme | Common Substrate | Signal Type | Product/Readout | Stop Solution |
|---|---|---|---|---|
| Horseradish Peroxidase (HRP) | TMB (Tetramethylbenzidine) | Colorimetric | Blue → Yellow; read at 450 nm [2] | Acid (e.g., H₂SO₄, HCl) [2] |
| Horseradish Peroxidase (HRP) | Hydrogen Peroxide + other chromogens | Colorimetric | Varies by chromogen | Acid [9] |
| Alkaline Phosphatase (AP) | pNPP (p-Nitrophenyl Phosphate) | Colorimetric | Yellow; read at 405 nm [9] | Base (e.g., NaOH) [2] |
| Alkaline Phosphatase (AP) | BCIP/NBT | Colorimetric | Blue precipitate [2] | Not Specified |
Experimental Protocols for ELISA Variants
The following protocols detail the setup of direct, indirect, and sandwich ELISAs, highlighting how the core components are utilized in each format.
Direct ELISA Protocol
The direct ELISA is the most straightforward format, using a single labeled antibody. [11]
- Coating: Dilute the antigen in a coating buffer (e.g., carbonate-bicarbonate buffer, pH 9.4) to a concentration of 2–10 µg/mL. Add 50–100 µL per well to a 96-well microplate and incubate for several hours to overnight at 4–37°C. [12] Wash the plate with PBS or a wash buffer to remove unbound antigen. [2]
- Blocking: Add a blocking buffer containing an irrelevant protein (e.g., 3–5% BSA in PBS) to all wells to cover any remaining protein-binding sites. Incubate for at least 1–2 hours at room temperature. Wash the plate. [9] [18]
- Detection with Conjugated Antibody: Add the enzyme-conjugated primary antibody specific to the antigen. Incubate for 1–2 hours at room temperature. Wash the plate thoroughly to remove any unbound antibody. [9]
- Signal Development and Reading: Add the appropriate enzyme substrate (e.g., TMB for HRP). Incubate in the dark for 15–30 minutes. Add the stop solution (e.g., 1M H₂SO₄ for TMB) and measure the absorbance immediately with a microplate reader. [9]
Indirect ELISA Protocol
The indirect ELISA introduces a secondary antibody for signal amplification, enhancing sensitivity. [16]
- Coating and Blocking: Perform the coating and blocking steps as described in the Direct ELISA protocol (Steps 1 and 2). [9]
- Primary Antibody Incubation: Add the unlabeled primary antibody specific to the antigen. Incubate for 1–2 hours at room temperature. Wash the plate. [9]
- Secondary Antibody Incubation: Add the enzyme-conjugated secondary antibody that is specific to the host species of the primary antibody (e.g., anti-rabbit IgG-HRP if the primary is from a rabbit). Incubate for 1–2 hours at room temperature. Wash the plate thoroughly. [9]
- Signal Development and Reading: Proceed with signal development and reading as in the Direct ELISA protocol (Step 4). [9]
Sandwich ELISA Protocol
The sandwich ELISA is the most sensitive and specific format, ideal for complex samples like serum or cell lysates. [11] [18] The protocol below is a detailed guide.
- Capture Antibody Coating: Dilute the capture antibody in a coating buffer to 1–10 µg/mL. Add 50–100 µL per well to a 96-well microplate. Cover the plate and incubate with gentle agitation for 2 hours at room temperature or overnight at 4°C. Wash the plate three times with wash buffer. [18]
- Blocking: Block the plate with a protein-based blocking buffer (e.g., PBS with 3–5% BSA) for at least 1–2 hours at room temperature to prevent non-specific binding. Wash the plate. [9] [18]
- Antigen Incubation: Add the samples, standards, and controls to the wells. Incubate for 90 minutes at 37°C to allow the antigen to bind to the capture antibody. Wash the plate to remove unbound material. [9] [18]
- Detection Antibody Incubation: Add the detection antibody specific to a different epitope on the antigen. This antibody can be enzyme-conjugated (direct detection) or unlabeled (requiring a subsequent step with a conjugated secondary antibody for indirect detection). Incubate for 1–2 hours at room temperature and wash. [9] [18]
- Signal Development and Reading: If an indirect detection method was used, add the enzyme-conjugated secondary antibody at this stage, incubate, and wash. [9] Finally, add the substrate, incubate, stop the reaction, and read the absorbance. [18]
Sandwich ELISA Workflow
Research Reagent Solutions
A successful ELISA relies on a suite of essential reagents and materials. The following table details key items and their functions for setting up a core ELISA laboratory.
Table 4: Essential Research Reagents and Materials for ELISA
| Item | Function / Description | Examples / Notes |
|---|---|---|
| Microplate | 96-well or 384-well plate serving as the solid phase. | Clear polystyrene for colorimetry; black/white for fluorescence/chemiluminescence. [12] |
| Coating Buffer | Buffer for diluting antigen or capture antibody for plate adsorption. | Carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4). [12] |
| Blocking Buffer | Solution of irrelevant protein to cover unused binding sites. | 1-5% BSA, ovalbumin, or casein in PBS to reduce background. [9] [18] |
| Wash Buffer | Buffer for removing unbound reagents between steps. | PBS or Tris buffer, often with a non-ionic detergent (e.g., Tween-20). [2] [9] |
| Antibody Pairs | Matched capture and detection antibodies for sandwich ELISA. | Must bind distinct, non-overlapping epitopes on the target antigen. [18] |
| Enzyme Conjugate | Antibody (or antigen) linked to the reporter enzyme. | HRP- or AP-conjugated primary or secondary antibodies. [2] [12] |
| Substrate | Chemical converted by the enzyme to a detectable signal. | TMB (HRP) or pNPP (AP) for colorimetric detection. [2] [9] |
| Stop Solution | Acidic or basic solution to halt the enzyme-substrate reaction. | 1-2 M H₂SO₄ or HCl; NaOH for some AP substrates. [2] |
| Microplate Reader | Instrument to measure the absorbance, fluorescence, or luminescence. | Spectrophotometer for colorimetric ELISA (read at 450nm for TMB). [2] |
Data Analysis and Best Practices
Accurate data analysis is critical for reliable quantification. Here are key pre- and post-assay considerations:
- Before Running the ELISA:
- Replicates: Run all standards and samples in duplicate or triplicate to assess pipetting error and calculate a coefficient of variation (CV). A CV of less than 20% is a best-practice target. [19]
- Standard Curve: Include a serial dilution of a known standard on every plate to account for inter-assay variability. [19]
- Controls: Use blank samples (buffer only) to subtract background absorbance and positive controls with known concentration to validate the assay. [19]
- Dilutions: Test samples at multiple dilutions to ensure at least one falls within the linear range of the standard curve. [19]
- After Running the ELISA:
- Standard Curve Fitting: Use a 4-parameter logistic (4-PL) curve fit for the standard dilution data, as this model typically provides the best fit for immunoassay data. [19]
- Background Subtraction: Subtract the average absorbance of the blank wells from all other readings. [19]
- Calculation: Use the standard curve to interpolate the concentration of unknown samples, remembering to multiply by any dilution factors applied. [19]
ELISA Data Analysis Steps
Enzyme-Linked Immunosorbent Assay (ELISA) detects and quantifies biological molecules by exploiting the potent catalytic activity of enzymes to generate an amplified, measurable signal from antigen-antibody interactions. This detection principle transforms an invisible molecular binding event into a quantifiable colorimetric, chemiluminescent, or fluorescent output, providing researchers with a powerful tool for precise protein and antibody measurement. The core of this system relies on enzymes conjugated to detection antibodies that catalyze the conversion of substrates into detectable products, with the signal intensity directly proportional to the target analyte concentration [9] [2].
Core Enzyme-Substrate Systems in ELISA
The specificity and sensitivity of ELISA detection depend on carefully matched enzyme-substrate pairs. The most common systems utilize Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP) due to their high turnover rates and stability when conjugated to antibodies [9] [2].
- Horseradish Peroxidase (HRP) catalyzes the reduction of hydrogen peroxide (H₂O₂) while oxidizing a chromogenic substrate. Tetramethylbenzidine (TMB) is a frequently used HRP substrate that produces a blue color during the reaction, which turns yellow when stopped with a strong acid like sulfuric or hydrochloric acid [9] [2]. The absorbance of this yellow solution is measured at 450 nm.
- Alkaline Phosphatase (AP) removes phosphate groups from its substrates. With para-Nitrophenylphosphate (pNPP) as a substrate, AP produces a yellow product, p-nitrophenol, which can be measured directly at 405-410 nm [9].
The remarkable signal amplification achieved stems from the enzyme's catalytic nature. A single enzyme molecule converts thousands of substrate molecules to product, greatly magnifying the signal from each antibody-antigen binding event [20]. For instance, carbonic anhydrase can turn over over 600,000 substrate molecules per second [20].
Detection Pathway Diagrams
The following diagrams illustrate the fundamental signaling pathways for the two primary enzyme systems used in ELISA.
Diagram Title: HRP-TMB Detection Pathway
Diagram Title: AP-pNPP Detection Pathway
Application Across ELISA Formats
The enzyme-substrate reaction is the universal detection endpoint across all major ELISA formats, though the methods for capturing the target and attaching the enzyme differ significantly. The following table compares how this detection principle is integrated into different assay architectures, each offering distinct advantages for specific applications [21] [9].
| ELISA Format | Assay Architecture | Integration of Enzyme-Substrate Detection | Key Advantages |
|---|---|---|---|
| Direct ELISA | Antigen is immobilized; detected by an enzyme-linked primary antibody. | Enzyme is directly conjugated to the primary antibody. Simplest workflow. | Minimal steps; avoids secondary antibody cross-reactivity. |
| Indirect ELISA | Antigen is immobilized; detected by an unlabeled primary antibody, then an enzyme-linked secondary antibody. | Enzyme is conjugated to a secondary antibody that binds the primary antibody. | Signal amplification through multiple secondary antibodies; highly sensitive; flexible. |
| Sandwich ELISA | Antigen is captured between a surface-bound antibody and a detection antibody. | Enzyme is typically linked via a secondary antibody that binds the detection antibody. | Highest specificity and sensitivity; suitable for complex samples. |
| Competitive ELISA | Sample antigen and labeled antigen compete for limited antibody binding sites. | Enzyme is conjugated to a reference antigen. Signal is inversely proportional to analyte concentration. | Ideal for detecting small molecules or inhibitors. |
Experimental Protocols and Performance Data
Recent research demonstrates the practical application of these detection principles, with sandwich ELISA being a preferred format for its high sensitivity and specificity. The following experimental protocols from current studies highlight the optimization of enzyme-substrate detection for specific targets.
Protocol 1: Sandwich ELISA for Pan-Merbecovirus Detection
A 2025 study established a sensitive sandwich ELISA for detecting the nucleocapsid (N) protein across multiple Merbecoviruses, including MERS-CoV and bat-derived viruses [22].
- Coating: Wells were pre-coated with a monoclonal capture antibody (1A8).
- Blocking: Blocked with a proprietary blocking buffer to prevent non-specific binding.
- Sample Incubation: Sample containing viral antigen was added and captured.
- Detection Antibody Binding: A biotinylated monoclonal detection antibody (10H6), recognizing a different epitope, was added.
- Enzyme Conjugate: Streptavidin conjugated to Horseradish Peroxidase (HRP) was used.
- Substrate Reaction: The HRP substrate TMB was added, producing a blue color.
- Signal Stop & Measurement: The reaction was stopped with H₂SO₄, and absorbance was read at 450 nm.
Performance Data: The assay demonstrated high sensitivity with limits of detection (LOD) below 7.81 ng/mL for all tested merbecoviruses, and as low as 1.25 ng/mL for VsCoV-1. It also detected infectious virus at 1.3 × 10³ PFU/mL, showcasing the power of enzyme-mediated signal amplification for sensitive antigen detection [22].
Protocol 2: Sandwich ELISA for Dengue Virus Envelope Protein
A 2025 study developed a sandwich ELISA for the envelope domain III (EDIII) protein of dengue virus type 2 (DENV-2) [23].
- Coating: Wells were coated with a rabbit polyclonal anti-DENV-2_EDIII antibody.
- Blocking: Blocking was performed with 5% skimmed milk powder.
- Sample Incubation: Serial dilutions of the purified DENV-2_EDIII antigen standard or test samples were added.
- Detection Antibody Binding: The same rabbit polyclonal anti-DENV-2_EDIII antibody was used for detection.
- Enzyme Conjugate: An HRP-conjugated goat anti-rabbit IgG secondary antibody was employed.
- Substrate Reaction: TMB substrate was added.
- Signal Stop & Measurement: The reaction was stopped with 2 M H₂SO₄, and absorbance was read at 450 nm.
Performance Data: This assay achieved an impressive LOD of 1.18 ng/mL and a quantitation range of 3.13–100 ng/mL, confirming that the ELISA format provides a wide dynamic range for accurate quantification [23].
The Scientist's Toolkit: Essential Reagents for ELISA Detection
Successful implementation of ELISA relies on a set of critical reagents, each playing a vital role in the assay's performance and the fidelity of the final enzyme-substrate signal.
| Reagent / Material | Function in Detection |
|---|---|
| Microplate | Solid polystyrene surface for immobilizing capture antibodies or antigens [2]. |
| Coating Antibody | High-affinity antibody that binds and immobilizes the target antigen to the plate (e.g., mouse mAb 1A8) [22]. |
| Blocking Buffer | Protein-based solution (e.g., BSA, skim milk) that covers unused plastic surface to prevent non-specific binding of detection reagents [24] [25]. |
| Detection Antibody | Antibody that binds to a distinct epitope on the captured antigen; may be biotinylated (e.g., mouse mAb 10H6) or directly enzyme-conjugated [22] [23]. |
| Enzyme Conjugate | Critical signal-generating component. Often a Streptavidin-HRP complex (binds biotinylated detection Ab) or an enzyme-linked secondary antibody [22] [24]. |
| Chromogenic Substrate | Molecule (e.g., TMB, pNPP) enzymatically converted into a colored, measurable product [9] [2]. |
| Stop Solution | Strong acid (e.g., H₂SO₄) or base that halts the enzyme-substrate reaction, stabilizing the signal for measurement [2]. |
| Plate Reader | Spectrophotometer that measures the optical density (absorbance) of the colored product in each well, typically at 450 nm for TMB or 405 nm for pNPP [2]. |
The enzyme-substrate reaction remains the cornerstone of ELISA detection, providing the critical link between specific molecular recognition and quantifiable signal output. Ongoing refinements in enzyme conjugates, substrate chemistry, and assay automation continue to push the boundaries of sensitivity and precision, ensuring ELISA remains an indispensable tool for researchers and clinicians in the life sciences.
The Enzyme-Linked Immunosorbent Assay (ELISA) is a foundational biochemical technique that leverages the specificity of antigen-antibody interactions to detect and quantify soluble substances such as peptides, proteins, antibodies, and hormones [12] [26]. Since its original description by Engvall and Perlmann in 1971, ELISA has become a gold standard in research and diagnostic laboratories worldwide due to its high sensitivity, specificity, and versatility [12] [9]. The core principle involves immobilizing an antigen on a solid surface and complexing it with an antibody linked to a reporter enzyme; detection is achieved by measuring the enzyme's activity upon substrate introduction [12].
This guide provides an objective comparison of the three major ELISA formats—Direct, Indirect, and Sandwich ELISA. It is designed to inform researchers, scientists, and drug development professionals in their selection of the most appropriate assay format for specific applications, supported by experimental data, detailed protocols, and key reagent solutions.
Core Principles and Methodologies
The General ELISA Workflow
All ELISA types share a common sequence of core steps, which ensures the specific binding and detection of the target analyte. The workflow below illustrates these fundamental stages.
Step 1: Plate Coating. The antigen or capture antibody is passively adsorbed onto the surface of a polystyrene microplate well through hydrophobic interactions [12] [9]. Coating is typically performed using a carbonate-bicarbonate buffer (pH 9.4) or phosphate-buffered saline (PBS), with incubation times ranging from several hours at 37°C to overnight at 4°C [12] [27].
Step 2: Blocking. After coating, any remaining unsaturated binding sites on the plastic surface are saturated with an irrelevant protein or molecule, such as Bovine Serum Albumin (BSA) or non-fat milk [12] [9]. This crucial step minimizes non-specific binding of detection antibodies in subsequent stages, thereby reducing background signal [28] [9]. Blocking is typically carried out for 1 to 2 hours at room temperature [27].
Step 3: Probing/Detection. The immobilized antigen is incubated with antigen-specific antibodies. The strategy for this step—whether using a labeled primary antibody or a matched set of unlabeled primary and labeled secondary antibodies—defines the type of ELISA and greatly influences its sensitivity and specificity [12] [29].
Step 4: Signal Measurement. A substrate specific to the reporter enzyme (e.g., HRP or AP) is added. The enzyme catalyzes a reaction that generates a measurable product, often a color change [2] [9]. The reaction is stopped with an acidic solution, and the optical density (OD) is measured with a microplate spectrophotometer, typically at 450 nm [30] [27]. The intensity of the signal is proportional to the amount of analyte in the sample.
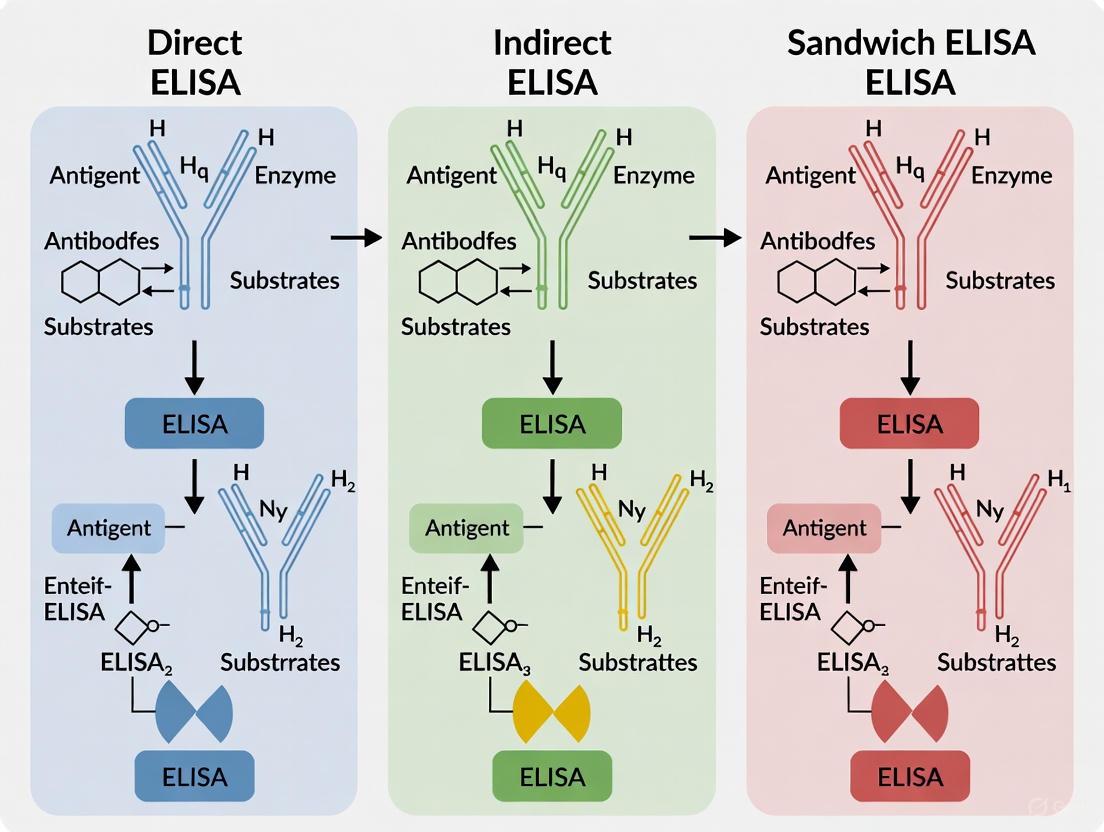
Comparative Workflows for Direct, Indirect, and Sandwich ELISA
The following diagram details the specific procedural and reagent differences between the three main ELISA types.
Comparative Analysis of ELISA Types
Detection Strategies and Performance
The table below summarizes the key characteristics, advantages, and disadvantages of each ELISA type to guide format selection.
Table 1: Comparison of Direct, Indirect, and Sandwich ELISA Methods
| Feature | Direct ELISA | Indirect ELISA | Sandwich ELISA |
|---|---|---|---|
| Principle | Antigen is immobilized; detected directly by a conjugated primary antibody [12] [29]. | Antigen is immobilized; detected by an unlabeled primary antibody, then a conjugated secondary antibody [12] [27]. | Antigen is captured by an immobilized antibody and detected by a second, specific antibody [12] [9]. |
| Key Steps | Coat antigen → Add conjugated primary Ab → Add substrate [9]. | Coat antigen → Add primary Ab → Add conjugated secondary Ab → Add substrate [9] [27]. | Coat capture Ab → Add antigen → Add detection Ab → (If needed: Add conjugated secondary Ab) → Add substrate [12] [9]. |
| Sensitivity | Lower, as there is no signal amplification [12] [29]. | High, due to signal amplification from multiple secondary antibodies binding to a single primary [12] [29]. | Highest, as the target antigen is bound by two specific antibodies, enhancing specificity and signal [12] [9]. |
| Specificity | High; reduced risk of cross-reactivity from secondary antibodies [12]. | Moderate; potential for cross-reactivity from the secondary antibody must be managed [12] [9]. | Very high; requires two distinct epitopes on the antigen to be bound, minimizing false positives [12] [26]. |
| Time Required | Faster (fewer steps) [12]. | Longer (extra incubation step) [12]. | Longest (multiple binding and incubation steps) [9]. |
| Cost & Flexibility | Higher cost and lower flexibility; each primary antibody must be individually conjugated [12]. | Lower cost and high flexibility; one conjugated secondary antibody can be used with many primary antibodies [12] [29]. | High cost; requires matched antibody pairs that recognize different epitopes on the same antigen [12] [9]. |
| Best For | Quick, initial screens; detecting immune responses in cells/tissues via immunohistochemistry [12]. | Antibody screening (e.g., serological tests), general protein detection, and when signal amplification is needed [2] [9]. | Quantifying complex samples with high precision (e.g., cytokines, hormones, biomarkers) [26] [9]. |
Quantitative Data Analysis and Interpretation
Accurate data analysis is critical for reliable quantification. The relationship between the measured Optical Density (OD) and analyte concentration differs by ELISA type.
- Standard Curve: Quantitative analysis requires a standard curve with known analyte concentrations, typically prepared via serial dilution [30]. The standard curve is plotted with concentration on the x-axis (log scale) and absorbance on the y-axis (linear scale) [2] [9].
- Signal-Concentration Relationship:
- In Sandwich and Indirect ELISA, the OD value is positively correlated with the analyte concentration [30]. More target analyte leads to more enzyme-conjugated antibody binding and a stronger signal.
- In Competitive ELISA (a variant not covered in detail here), the relationship is inverse; a higher sample analyte concentration results in a lower signal [12] [30].
- Curve Fitting: The standard curve is typically fitted using a 4-parameter logistic (4PL) or 5-parameter logistic (5PL) model, which accurately captures the sigmoidal nature of the assay's response across a wide dynamic range [30]. The coefficient of determination (R²) should be >0.98 to validate the curve fit [30]. Sample concentrations are then interpolated from this curve.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of an ELISA requires specific, high-quality reagents and equipment. The following table details the essential components for setting up an ELISA laboratory.
Table 2: Essential Research Reagent Solutions and Materials for ELISA
| Item | Function / Description | Examples / Notes |
|---|---|---|
| Solid Phase | 96-well or 384-well polystyrene microplates that passively bind proteins [12] [2]. | Clear plates for colorimetry; white/black for fluorescence/chemiluminescence. Not tissue culture treated [12]. |
| Coating Buffer | Alkaline buffer for diluting antigen/antibody for plate coating to facilitate adsorption [12] [27]. | Carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4) [12] [27]. |
| Blocking Buffer | A solution of irrelevant protein to cover any unsaturated binding sites on the plate to prevent non-specific binding [12] [9]. | 1-5% BSA, non-fat dry milk, or other animal proteins in PBS or Tris buffer [28] [9] [27]. |
| Capture & Detection Antibodies | High-affinity, specific antibodies are the backbone of any ELISA. For sandwich ELISA, a "matched pair" is required [12] [31]. | Must be validated for ELISA. Capture and detection antibodies should be from different host species to avoid interference [12]. |
| Enzyme Conjugates | Reporter enzymes linked to the primary or secondary antibody for signal generation [12] [2]. | Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP) are most common [12] [9]. |
| Enzyme Substrate | The compound acted upon by the enzyme to produce a detectable signal [2] [9]. | TMB (turns blue with HRP, then yellow when stopped) and pNPP (turns yellow with AP) are common chromogenic substrates [2] [9]. |
| Wash Buffer | A buffered solution with a mild detergent used to remove unbound reagents between steps [2] [27]. | PBS or Tris buffer with 0.01-0.1% Tween-20 (PBST/TBST) [28] [27]. |
| Stop Solution | An acidic or basic solution to halt the enzyme-substrate reaction at a defined timepoint [2]. | 1M or 2M H₂SO₄ is commonly used for HRP/TMB reactions [2] [27]. |
| Microplate Reader | An instrument that measures the optical density (OD) in each well of the plate [2] [30]. | Spectrophotometer for colorimetric detection; can also be configured for fluorescent or luminescent readouts [30] [31]. |
Troubleshooting Common Experimental Issues
Even well-optimized ELISAs can encounter problems. The table below outlines common issues and their solutions.
Table 3: Common ELISA Problems and Troubleshooting Strategies
| Problem | Potential Causes | Solutions |
|---|---|---|
| High Background Signal | Incomplete plate washing [28].Inadequate blocking [28].Excessive detection antibody concentration [30] [28]. | Increase number of wash cycles; add a short incubation soak during washes [28].Increase blocking buffer concentration (e.g., 1% to 2% BSA) or extend blocking time [28].Titrate the detection antibody to find the optimal dilution [30]. |
| Low or No Signal | Expired or degraded substrate (e.g., TMB) [30].Inaccurate reagent preparation or missing a key reagent [30].Antigen concentration below assay detection limit. | Use fresh substrate and ensure it is stored correctly [30].Double-check protocol and pipetting. Ensure all reagents are added in the correct order [30].Concentrate the sample or use a more sensitive ELISA format (e.g., switch from direct to indirect) [29]. |
| High Variation Between Replicates | Inconsistent pipetting [30].Inhomogeneous samples or reagents [30].Plate edge effects (evaporation). | Calibrate pipettes and ensure proper pipetting technique [30].Vortex samples and centrifuge briefly before use to gather liquid [30].Use a plate seal during incubations and avoid using outer wells if necessary. |
The choice between Direct, Indirect, and Sandwich ELISA is a strategic decision that depends on the specific experimental goals and constraints.
- Direct ELISA offers simplicity and speed, making it suitable for quick antigen detection when a conjugated primary antibody is available and high sensitivity is not the primary concern [12] [29].
- Indirect ELISA provides enhanced sensitivity through signal amplification and greater flexibility, making it ideal for antibody screening applications and when the same secondary antibody can be used across multiple assays [12] [9].
- Sandwich ELISA delivers the highest specificity and sensitivity for antigen quantification, indispensable for measuring low-abundance proteins in complex biological mixtures like serum or cell culture supernatants, despite requiring more resources and optimization [12] [26] [9].
Understanding the principles, advantages, and limitations of each format empowers researchers to select the most appropriate tool for their needs, thereby ensuring robust, reliable, and meaningful experimental results in both basic research and drug development.
Methodology in Action: Step-by-Step Protocols and Applications
The Enzyme-Linked Immunosorbent Assay (ELISA) is a foundational technique in biomedical research and clinical diagnostics, enabling the detection and quantification of specific proteins, antibodies, hormones, and other biomolecules within complex mixtures [12]. Among its various formats, the direct ELISA represents the most straightforward approach, characterized by its simplified protocol and minimal procedural steps. In this assay, the antigen is immobilized directly onto a microplate well and detected using a single primary antibody that is conjugated directly to a reporter enzyme [12] [11]. This guide deconstructs the direct ELISA protocol, compares its performance against other common ELISA variants, and provides the visual tools necessary for effective experimental planning and execution.
Visualizing the Direct ELISA Workflow
The following diagram illustrates the fundamental steps and molecular interactions in a direct ELISA.
Detailed Step-by-Step Direct ELISA Protocol
Plate Coating: Antigen Immobilization
Procedure: Dilute the purified antigen in a coating buffer. Carbonate-bicarbonate buffer (pH 9.6) or phosphate-buffered saline (PBS, pH 7.4) are commonly used [12] [32]. Add 50-100 µL of the antigen solution to each well of a 96-well polystyrene microplate [27]. Cover the plate with adhesive plastic to prevent evaporation and incubate for 1 hour at 37°C or overnight at 4°C [32]. The optimal antigen concentration for coating should be determined experimentally but is generally below 20 µg/mL to avoid high background [27].
Mechanism: This step relies on passive adsorption, where hydrophobic interactions between non-polar protein residues and the plastic surface result in irreversible immobilization of the antigen [12].
Blocking: Minimizing Non-Specific Binding
Procedure: Remove the coating solution by flicking the plate over a sink. Add 200-300 µL of blocking buffer per well. Common blocking agents include 1-5% Bovine Serum Albumin (BSA), non-fat dry milk, or animal sera in PBS [27] [9]. Incubate for 1-2 hours at room temperature. Wash the plate once with PBS containing 0.05% Tween 20 (wash buffer) [32].
Mechanism: Blocking agents saturate any remaining protein-binding sites on the polystyrene surface that were not occupied during the coating step, thereby preventing non-specific binding of detection antibodies in subsequent steps and reducing background signal [9].
Detection: Antibody Binding
Procedure: Prepare the enzyme-conjugated primary antibody in blocking buffer at the manufacturer's recommended dilution (typically 1-10 µg/mL) [12]. Add 100 µL of the antibody solution to each well. Incubate for 1-2 hours at room temperature with gentle shaking. Remove the antibody solution and wash the plate 3-5 times with wash buffer (300 µL per well) to remove unbound antibodies [27] [32].
Mechanism: The conjugated primary antibody specifically binds to epitopes on the immobilized antigen. Common enzyme labels include Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP), which will later catalyze the conversion of substrates into detectable products [12].
Signal Development and Detection
Procedure: Add 100 µL of substrate solution to each well. For HRP-conjugated antibodies, use TMB (3,3',5,5'-tetramethylbenzidine), which produces a blue color, or ABTS (2,2'-azino-di-[3-ethyl-benzothiazoline-6-sulfonic acid] diammonium salt), which produces a green color. For AP-conjugated antibodies, PNPP (p-Nitrophenyl Phosphate) is commonly used, yielding a yellow product [33] [34]. Incubate for 15-30 minutes at room temperature, protected from light. Stop the reaction when sufficient color develops (for TMB, add an equal volume of stop solution, typically 0.16 M sulfuric acid, which changes the color to yellow) [32]. Measure the absorbance immediately using a microplate reader at the appropriate wavelength (450 nm for acid-stopped TMB, 405 nm for PNPP) [33].
Mechanism: The enzyme conjugated to the detection antibody catalyzes the conversion of the substrate into a colored, fluorescent, or chemiluminescent product. The intensity of the signal is directly proportional to the amount of antigen present in the well [12].
Comparative Analysis of ELISA Formats
Performance Characteristics of ELISA Types
Table 1: Comparative analysis of direct, indirect, sandwich, and competitive ELISA formats
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Sensitivity | Low to moderate [12] [11] | High (signal amplification) [12] [9] | Highest (typically 2-5x more sensitive than direct/indirect) [33] [11] | Moderate [11] |
| Time Required | ~3-4 hours (fastest protocol) [12] | ~4-5 hours (additional incubation step) [12] | ~5-6 hours (longest protocol) [11] | ~4-5 hours [11] |
| Complexity | Low (fewest steps) [12] | Moderate [12] | High (requires antibody pairing) [12] [11] | Moderate [11] |
| Specificity | Lower (single antibody) [11] | Moderate (potential for secondary cross-reactivity) [12] [9] | Highest (two antibodies required) [12] [11] | Lower (single antibody) [11] |
| Cost | Higher (labeled primary antibodies needed) [9] | Lower (versatile secondary antibodies) [12] [9] | Highest (two specific antibodies) [12] | Moderate [11] |
| Signal Amplification | No amplification (minimal signal) [12] | Yes (multiple secondary antibodies per primary) [12] | Yes (can be combined with indirect detection) [12] | No amplification [11] |
| Antigen Requirements | Must be adsorbable to plate [12] | Must be adsorbable to plate [12] | Must have at least two epitopes [33] | Suitable for small antigens [12] [11] |
| Typical Applications | Antibody affinity/specificity testing [11] | Measuring endogenous antibodies [11] | Quantifying biomarkers in complex samples [11] | Measuring small molecules/hormones [12] [11] |
Visual Comparison of ELISA Methodologies
The following diagram illustrates the key structural and procedural differences between the four main ELISA formats.
The Researcher's Toolkit: Essential Reagents and Materials
Table 2: Key research reagent solutions for direct ELISA
| Reagent/Material | Function | Typical Composition/Examples |
|---|---|---|
| Microplate | Solid phase for immobilization | 96-well polystyrene plates (clear for colorimetric detection) [12] |
| Coating Buffer | Facilitates antigen adsorption | 50 mM carbonate-bicarbonate buffer (pH 9.6) or PBS (pH 7.4) [27] [32] |
| Blocking Buffer | Prevents non-specific binding | 1-5% BSA, non-fat dry milk, or animal serum in PBS [27] [9] |
| Wash Buffer | Removes unbound reagents | PBS or Tris-buffered saline with 0.05% Tween 20 detergent [32] [35] |
| Enzyme-Conjugated Primary Antibody | Specific detection | HRP or AP conjugated to antigen-specific antibody [12] |
| Enzyme Substrate | Generates detectable signal | TMB, ABTS (for HRP); PNPP (for AP) [33] [34] |
| Stop Solution | Terminates enzyme reaction | 0.16-2 M sulfuric acid (for TMB); 2 N NaOH (for PNPP) [33] [32] |
Applications and Strategic Implementation
When to Choose Direct ELISA
The direct ELISA format offers distinct advantages in specific research scenarios. Its streamlined protocol makes it ideal for high-throughput screening applications where speed is prioritized over maximal sensitivity [12]. The method is particularly valuable for assessing antibody affinity and specificity, as it eliminates potential cross-reactivity from secondary antibodies [11]. Additionally, direct detection is commonly employed in immunohistochemical staining of tissues and cells, where the direct labeling approach provides precise localization [12].
Limitations and Considerations
Researchers should recognize the inherent limitations of direct ELISA. The technique generally exhibits lower sensitivity compared to indirect or sandwich formats due to the absence of signal amplification [12]. The requirement for individually conjugated primary antibodies makes assay development time-consuming and expensive [12]. Additionally, the labeling process of primary antibodies with reporter enzymes may potentially affect their immunoreactivity, compromising binding affinity in some cases [12].
The direct ELISA remains an essential technique in the molecular biologist's arsenal, particularly when experimental priorities include protocol simplicity, rapid results, and elimination of secondary antibody cross-reactivity. While it may not offer the sensitivity of sandwich ELISA or the versatility of indirect ELISA, its straightforward approach makes it invaluable for specific applications including initial antibody characterization and high-throughput screening. Understanding the fundamental workflow, as visualized in this guide, along with its performance characteristics relative to other immunoassay formats, enables researchers to make informed methodological selections based on their specific experimental requirements, sample availability, and detection sensitivity needs.
The Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technique in immunology and molecular biology for detecting and quantifying specific proteins or antigens [27] [12]. Since its initial description in 1971, it has evolved into a safer, more convenient alternative to radioimmunoassay and has become indispensable for both research and clinical diagnostics [36]. Among its various formats, the indirect ELISA is particularly renowned for its superior sensitivity and flexibility, making it a preferred choice for applications like quantifying antibody responses in serological studies and vaccine trials [37] [11].
This guide provides an objective comparison of the indirect ELISA against other common variants—direct, sandwich, and competitive ELISA—framed within a broader thesis on ELISA technology. We will delve into the detailed protocol, explore advanced signal amplification strategies, and summarize key performance data to help researchers and drug development professionals select the optimal assay for their specific needs.
Core Principles: Indirect vs. Other ELISA Formats
The fundamental principle of any ELISA is the immobilization of an antigen on a solid surface (typically a microplate) and its detection via antibodies linked to a reporter enzyme, which generates a measurable signal [27] [12]. The formats differ primarily in their approach to capture and detection.
The table below summarizes the core characteristics of the four main ELISA types:
| ELISA Type | Core Principle | Key Advantages | Key Disadvantages | Ideal Applications |
|---|---|---|---|---|
| Direct ELISA [38] [11] | A labeled primary antibody binds directly to the immobilized antigen. | - Quick and simple protocol [12] [39].- Fewer steps, lower risk of cross-reactivity [29] [12]. | - Less sensitive (no signal amplification) [12] [36].- Requires conjugated primary antibodies [12]. | - Assessing antibody affinity [11].- Rapid antigen screening [38]. |
| Indirect ELISA [38] [11] | An unlabeled primary antibody binds the antigen; a labeled secondary antibody then binds the primary. | - High sensitivity (signal amplification) [29] [12].- Flexible (same secondary can detect many primaries) [12] [11].- Maximum immunoreactivity of primary antibody [12]. | - More complex, longer protocol [39].- Potential for secondary antibody cross-reactivity [12] [11]. | - Measuring endogenous antibodies (e.g., serology) [36] [11].- Antibody titration in vaccine studies [38]. |
| Sandwich ELISA [38] [11] | Two antibodies specific to different epitopes "sandwich" the antigen. The detection can be direct or indirect. | - Highest specificity and sensitivity [12] [11].- Compatible with complex samples (e.g., serum) [11]. | - Requires a matched pair of antibodies [38].- Technically demanding and costly to develop [38]. | - Quantifying specific antigens in complex samples [38].- Biomarker detection in disease diagnostics [38]. |
| Competitive/Inhibition ELISA [12] [38] | Sample antigen and a labeled reference antigen compete for a limited number of antibody binding sites. | - Useful for quantifying small molecules [38] [11].- Flexible, can be adapted from other formats [38]. | - Less sensitive than sandwich ELISA [38].- Requires careful optimization [38]. | - Detecting small molecules (drugs, hormones) [38] [36].- Screening for contaminants [38]. |
Visualizing the Indirect ELISA Workflow
The following diagram illustrates the key steps and reagent interactions in a typical indirect ELISA procedure.
Detailed Indirect ELISA Protocol and Reagent Solutions
The following protocol is adapted from optimized methodologies used in recent research, such as for quantifying virus-specific antibodies [37].
Sample Preparation
Samples like serum, plasma, or cell culture supernatants must be properly prepared. For serum/plasma, collect blood in an appropriate anti-coagulant (e.g., EDTA), centrifuge at 1,000–10,000 x g for 10 minutes at 4°C, and carefully collect the supernatant [27]. Aliquot to minimize freeze-thaw cycles and store at -80°C [27]. Before the assay, heat-inactivate serum or plasma at 56°C for 30 minutes to denature complement proteins, then centrifuge at 1,000 x g for 10 minutes to remove precipitates [37].
Step-by-Step Protocol
- Plate Coating: Dilute the purified antigen in a carbonate-bicarbonate coating buffer (pH ~9.4) to a concentration generally below 20 µg/mL to avoid high background [27]. Add 100 µL per well to a medium-binding 96-well microplate. Cover the plate and incubate with gentle agitation for 2 hours at room temperature or overnight at 4°C [27] [37].
- Washing: Flick the plate to remove the coating solution. Wash each well three times with a wash buffer, such as PBS containing 0.05% Tween-20 (PBST). If washing manually, firmly tap the plate upside down on absorbent paper after each wash to remove residual liquid [27] [37].
- Blocking: Add 200 µL of a blocking buffer to each well to cover all unsaturated binding sites. Common blockers include 1% Bovine Serum Albumin (BSA) in PBST or commercially available casein blockers [27] [37]. Incubate with gentle agitation for 1–2 hours at room temperature.
- Primary Antibody Incubation: Dilute the primary antibody (e.g., patient serum) in a suitable diluent like BSA or goat serum assay buffer [37]. Add 100 µL of the diluted sample or standard to the wells. Cover the plate and incubate for 2 hours at room temperature or overnight at 4°C [27].
- Washing: Wash the plate as in step 2, repeating the process three times to remove unbound primary antibody.
- Secondary Antibody Incubation: Dilute an enzyme-conjugated secondary antibody (e.g., Goat anti-human IgG-HRP) in blocking buffer. Add 100 µL per well, cover the plate, and incubate for 2 hours at room temperature [27] [37].
- Washing: Perform a final wash step, again repeating three times to ensure all unbound secondary antibody is removed.
- Signal Detection: Add 100 µL of an appropriate substrate to each well. For Horseradish Peroxidase (HRP), Tetramethylbenzidine (TMB) is a common substrate. Incubate for 10-30 minutes at room temperature in the dark [37].
- Stop and Read: Stop the enzymatic reaction by adding 50-100 µL of a stop solution (e.g., 1M H₂SO₄ or a commercial TMB stop solution) [37]. Measure the absorbance immediately using a plate reader at the appropriate wavelength (e.g., 450 nm for TMB).
The Researcher's Toolkit: Essential Reagents
This table details the key reagents required for a successful indirect ELISA.
| Reagent / Solution | Function / Role | Example / Composition |
|---|---|---|
| Coating Buffer | Provides optimal pH and ionic conditions for antigen adsorption to the plate. | Carbonate-Bicarbonate buffer (pH 9.4) [27] |
| Wash Buffer | Removes unbound reagents and reduces non-specific background signal. | PBS (Phosphate Buffered Saline) + 0.05% Tween-20 (PBST) [37] |
| Blocking Buffer | Covers any remaining protein-binding sites on the plate to prevent non-specific antibody binding. | 1% BSA in PBST, 5% Goat Serum, or Commercial Casein Buffer [27] [37] |
| Primary Antibody | Binds specifically to the target antigen of interest. | Patient serum, monoclonal antibody, or polyclonal antiserum [36] |
| Enzyme-Linked Secondary Antibody | Binds to the primary antibody and produces a measurable signal. Must be specific to the host species of the primary antibody. | Goat anti-human IgG-HRP, Donkey anti-rabbit IgG-AP [27] [37] |
| Enzyme Substrate | Reacts with the enzyme on the secondary antibody to generate a detectable (e.g., colorimetric) product. | TMB (for HRP), pNPP (for Alkaline Phosphatase) [37] |
Advanced Considerations: Signal Amplification and Optimization
Signal Amplification Strategies
The inherent signal amplification in indirect ELISA comes from multiple secondary antibodies binding to a single primary antibody [12]. For detecting low-abundance targets, this can be further enhanced using biotin-streptavidin chemistry [36].
In this advanced format, a biotin-conjugated primary or secondary detection antibody is used. This is followed by the addition of streptavidin conjugated to HRP (SA-HRP) or a poly-HRP-40 conjugate, which can bind multiple biotin molecules, dramatically increasing the number of enzyme molecules per antibody-antigen complex and thus the assay's sensitivity [37]. The diagram below illustrates this powerful amplification system.
Key Optimization Parameters
- Antibody Titration: The optimal dilution for both primary and secondary antibodies must be determined empirically through checkerboard titration to maximize the signal-to-noise ratio [27] [12].
- Specificity and Cross-Reactivity: A major consideration in indirect ELISA is ensuring the secondary antibody is specific only to the primary antibody and does not cross-react with the capture antigen or other components [12] [36]. Using cross-adsorbed secondary antibodies and appropriate blocking buffers is critical [12].
- Sample Matrix Effects: Complex sample matrices like serum can interfere with antibody binding. Using a sample diluent that matches the matrix of the standard curve (e.g., containing a percentage of the relevant serum) can improve accuracy.
Comparative Performance Data and Future Trends
Quantitative Performance Comparison
The table below summarizes objective performance metrics across ELISA formats, illustrating the trade-offs between different methods.
| Performance Metric | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Assay Time | ~3-4 hours (Fastest) [11] | ~4-6 hours (Moderate) [39] | ~5-8 hours (Longest) [39] | ~4-6 hours (Moderate) |
| Sensitivity | Low (No amplification) [29] [36] | High (Amplification via secondary Ab) [29] [12] | Very High (Amplification & two antibodies) [11] | Moderate (Limited by competition) [38] |
| Specificity | Moderate (Single antibody) [11] | Moderate (Depends on secondary) [11] | Very High (Two distinct antibodies) [12] [11] | Moderate (Single antibody) [11] |
| Flexibility | Low (Each primary must be conjugated) [12] | High (Same secondary for many primaries) [12] [11] | Low (Requires matched pair) [38] | Moderate [38] |
| Cost & Complexity | Low (Fewer steps/reagents) | Low to Moderate | High (Two specific antibodies) [38] | Moderate to High |
The Future: Next-Generation ELISA
The ELISA market is evolving, with a significant shift toward "Next-Generation ELISA" or ELISA 2.0 platforms [40]. Key trends include:
- Multiplexing: Technologies like Luminex and Meso Scale Discovery (MSD) enable the simultaneous quantification of multiple analytes from a single sample, revolutionizing biomarker profiling [40] [38].
- Advanced Detection Methods: The replacement of traditional colorimetric substrates with chemiluminescent, electrochemical, and fluorescent reporters offers superior quantification and sensitivity, crucial for detecting low-abundance biomarkers in early disease diagnosis [40].
- Digital ELISA and Automation: Digital ELISA platforms push sensitivity to the single-molecule level, while integrated automation and microfluidic "lab-on-a-chip" devices enhance throughput, reproducibility, and reduce costs [40].
The indirect ELISA remains a powerful and versatile tool in the scientific arsenal, striking an excellent balance between sensitivity, flexibility, and practicality. Its core strength lies in its signal amplification mechanism, making it ideally suited for quantifying antibody levels in serology, immunology research, and vaccine development.
The choice of ELISA format, however, is fundamentally dictated by the experimental question and available reagents. For maximum specificity and sensitivity for an antigen, a sandwich ELISA is superior. For small molecules, a competitive format is necessary. For straightforward antigen detection where sensitivity is not paramount, a direct ELISA may suffice.
As immunoassay technology advances, the principles of the indirect ELISA continue to be refined and integrated into more powerful, multiplexed, and automated systems, ensuring its continued relevance in both basic research and clinical diagnostics.
The sandwich ELISA (Enzyme-Linked Immunosorbent Assay) is a powerful plate-based immunoassay technique renowned for its high sensitivity and specificity in detecting and quantifying target antigens within complex biological mixtures [41] [9]. This method earns its "sandwich" nomenclature from the characteristic structure formed during the assay: the target antigen is bound between two highly specific antibodies—a capture antibody and a detection antibody [12]. This dual-antibody recognition system significantly reduces cross-reactivity and enhances specificity compared to other ELISA formats, making it the format of choice for detecting low-abundance proteins in samples like serum, plasma, or cell lysates [18] [42].
The fundamental principle relies on the use of two antibodies that recognize different, non-overlapping epitopes on the target antigen [43]. Initially, a capture antibody is immobilized onto a solid-phase microplate. When the sample is added, the target antigen binds to this fixed antibody. Following a wash step to remove unbound materials, a detection antibody is introduced, which binds to a different epitope on the now-immobilized antigen, completing the "sandwich" complex [41] [12]. The detection antibody is typically conjugated to an enzyme, such as Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP), which, upon addition of a substrate, generates a measurable signal proportional to the amount of captured antigen [9] [12].
Comparative Analysis of ELISA Formats
While the sandwich ELISA is highly effective, understanding its performance relative to other common formats is crucial for selecting the appropriate method. The table below provides a structured comparison of direct, indirect, and sandwich ELISA types.
Table 1: Performance Comparison of Major ELISA Types
| Feature | Direct ELISA | Indirect ELISA | Sandwich ELISA |
|---|---|---|---|
| Sensitivity | Low (minimal signal amplification) [12] | High (signal amplification via secondary antibody) [12] | Highest (two antibodies enhance specificity and sensitivity) [9] [12] |
| Specificity | High (only one antibody used) [42] | Moderate (risk of secondary antibody cross-reactivity) [9] [12] | Highest (requires two distinct epitopes for detection) [42] |
| Time Required | Quickest (fewer steps) [12] | Moderate (additional incubation step) [12] | Longest (multiple incubation and wash steps) [9] |
| Complexity & Cost | Low | Moderate | High (requires matched antibody pair) [9] |
| Antigen Requirements | Must be able to adsorb to plate [12] | Must be able to adsorb to plate [9] | Must be large enough to have at least two non-overlapping epitopes [42] |
| Key Advantage | Speed, minimizes cross-reactivity [12] | Signal amplification, wide availability of labeled secondary antibodies [12] | Superior sensitivity and specificity for complex samples [12] [42] |
| Primary Disadvantage | Lower sensitivity, requires labeling of every primary antibody [12] | Potential for cross-reactivity from secondary antibody [9] | Time-consuming and requires optimized, matched antibody pairs [9] [12] |
Recent experimental data further underscores the performance of the sandwich ELISA principle in specific applications. A 2025 study evaluating serological assays for SARS-CoV-2 antibodies in animals found that a commercial competitive ELISA (ELISA-1), which is based on the sandwich principle and targets the Receptor Binding Domain (RBD), demonstrated superior diagnostic performance. It exhibited the highest sensitivity for detecting seropositive animals compared to another RBD-targeting assay and a nucleoprotein-targeting double antigen sandwich assay (ELISA-3) [44]. This highlights that the choice of target antigen within a format is also a critical factor for performance.
Detailed Sandwich ELISA Protocol
This section provides a comprehensive, step-by-step protocol for a colorimetric sandwich ELISA, adaptable for chemiluminescent detection with noted modifications [41] [18] [43].
Reagent and Material Preparation
The following reagents are essential for performing a sandwich ELISA. Proper preparation is critical for assay success.
Table 2: Key Research Reagent Solutions for Sandwich ELISA
| Reagent/Material | Function/Description | Example Formulation / Notes |
|---|---|---|
| Microplate | Solid surface for antibody immobilization and reaction vessel. | Clear 96-well plate for colorimetry; white/opaque for chemiluminescence [41] [12]. |
| Coating Buffer | Medium for diluting and immobilizing the capture antibody on the plate. | 50 mM carbonate-bicarbonate buffer, pH 9.4 [41] [43]. |
| Capture Antibody | Binds and immobilizes the target antigen to the plate. | Monoclonal or affinity-purified polyclonal, diluted in coating buffer (e.g., 1-10 µg/mL) [41] [18]. |
| Blocking Buffer | Covers any remaining protein-binding sites to prevent non-specific binding. | PBS or TBS with 1-5% BSA or a proprietary blocking agent [41] [18]. |
| Wash Buffer | Removes unbound reagents and sample matrix between steps. | PBS or TBS with 0.05% Tween 20 [41] [43]. |
| Detection Antibody | Binds to a different epitope on the captured antigen. | Specific antibody conjugated to HRP/AP, or a biotinylated antibody [41] [18]. |
| Enzyme Conjugate | Required if detection antibody is biotinylated. | Streptavidin-HRP, diluted in blocking buffer (e.g., 1:5,000) [41]. |
| Substrate | Converted by the enzyme to a detectable signal. | TMB (colorimetric, HRP) or Pico Chemiluminescent substrate (chemiluminescent, HRP) [41]. |
| Stop Solution | Halts the enzyme-substrate reaction. | 0.16 M sulfuric acid (for TMB) [41]. |
| Microplate Reader | Measures the absorbance or luminescence of each well. | Spectrophotometer (450 nm for TMB) or luminometer [41]. |
Step-by-Step Workflow
The diagram below illustrates the core procedural workflow and the molecular interactions at each stage of the sandwich ELISA process.
Sandwich ELISA Procedural Workflow
The procedural steps are as follows:
Plate Coating: Dilute the capture antibody in a coating buffer to a concentration typically between 1–10 µg/mL [43]. Add 100 µL of this solution to each well of a microplate. Cover the plate and incubate for 1 hour at room temperature or overnight at 2–8°C for optimal binding [41]. After incubation, aspirate the solution and wash the plate once with >300 µL of wash buffer. Invert and tap the plate on absorbent paper to remove excess liquid [41].
Blocking: Add 300 µL of blocking buffer per well to cover all remaining protein-binding sites on the plastic surface. Incubate for 1 hour at room temperature [41]. Aspirate the blocking buffer and tap the plate to remove excess liquid. This step is crucial for minimizing non-specific background signal [9].
Sample and Antigen Incubation: Prepare serial dilutions of your protein standard in blocking buffer. Similarly, prepare dilutions of your test samples [18]. Add 100 µL of each standard (in duplicate) and sample into designated wells. Incubate for 1–2 hours at room temperature with gentle continual shaking [41].
Washing: Aspirate the liquid from the wells. Wash each well with >300 µL of wash buffer. Invert and tap the plate to remove excess liquid. Repeat this wash process five times to ensure all unbound substances are thoroughly removed [41].
Detection Antibody Incubation: Prepare the detection antibody solution by diluting it in blocking buffer according to the manufacturer's instructions [41]. Add 100 µL of this solution to each well. Incubate for 2 hours at room temperature with gentle shaking. Aspirate and wash the plate five times, as in Step 4 [41].
- Note: If the detection antibody is directly conjugated to HRP, proceed to Step 7.
Enzyme Conjugate Incubation (if needed): If a biotinylated detection antibody is used, prepare a working solution of Streptavidin-HRP in blocking buffer (e.g., at a 1:5,000 dilution). Add 100 µL to each well and incubate for 1 hour at room temperature with shaking [41]. Aspirate and perform another five wash steps.
Signal Detection and Measurement: Add 100 µL of substrate solution (e.g., TMB for HRP) to each well. Incubate for 5-30 minutes at room temperature, protected from light, until the desired color intensity develops [41]. Stop the reaction by adding 100 µL of stop solution (e.g., 0.16 M sulfuric acid for TMB), which will change the color from blue to yellow [41]. Measure the absorbance at 450 nm within 30 minutes of stopping the reaction using a microplate reader [41].
Troubleshooting and Best Practices
Successful implementation of sandwich ELISA requires attention to critical factors. A primary challenge is the selection of a matched antibody pair, where the capture and detection antibodies must bind to distinct, non-overlapping epitopes on the target antigen to avoid steric hindrance [18] [43]. Furthermore, the target antigen must possess at least two antibody-binding sites, making this format less suitable for small molecules or haptens [42].
For quantitative analysis, include a standard curve with known concentrations of purified antigen in every assay run. The absorbance data can be analyzed using log-log or four-parameter curve fitting to interpolate sample concentrations [41]. For assay validation, parameters such as sensitivity, specificity, and precision (measuring intra-assay and inter-assay coefficient of variation) should be established [45]. Recent studies demonstrate that in-house developed ELISA methods, when properly validated, can achieve performance metrics comparable to commercial kits, with sensitivities and specificities exceeding 97% [45], providing a cost-effective solution for large-scale surveillance studies [46].
The Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technique in immunological research and clinical diagnostics, enabling the detection and quantification of specific biomolecules. Among its various formats, Direct ELISA offers a streamlined approach particularly well-suited for antigen screening and immune response analysis. This technique's simplicity and efficiency make it invaluable for researchers and drug development professionals who require rapid, initial screening of antigen presence and immune reactivity. As part of a broader comparison of ELISA methodologies, this review examines the specific applications, advantages, and limitations of Direct ELISA, providing experimental protocols and performance data to guide researchers in selecting the appropriate assay format for their specific needs.
Principles of Direct ELISA
Direct ELISA operates on a fundamental principle of antigen-antibody interaction, where the target antigen is immobilized directly onto a microplate surface and detected using a single enzyme-conjugated primary antibody [11] [47]. This direct detection method contrasts with other ELISA formats that incorporate additional amplification steps. The assay begins with antigen adsorption to the solid phase, typically a 96-well polystyrene plate, through passive adsorption mediated by hydrophobic interactions between non-polar protein residues and the plastic surface [12]. Following coating, any remaining binding sites on the plate are blocked with irrelevant proteins such as bovine serum albumin (BSA) to prevent nonspecific binding [9].
The key differentiator of Direct ELISA is the subsequent addition of a primary antibody that is directly conjugated to an enzyme reporter, such as horseradish peroxidase (HRP) or alkaline phosphatase (AP) [12]. After incubation and washing to remove unbound antibody, an appropriate substrate is added that produces a measurable color change when catalyzed by the enzyme [2]. The intensity of this signal is directly proportional to the amount of antigen present in the sample, allowing for quantification when compared to a standard curve [48]. This straightforward workflow—antigen coating, blocking, conjugated antibody incubation, and substrate addition—makes Direct ELISA one of the most efficient immunoassay formats available.
Comparative Analysis of ELISA Formats
The selection of an appropriate ELISA format depends on various factors including the research objective, sample complexity, required sensitivity, and available reagents. The table below provides a comprehensive comparison of the four principal ELISA types:
Table: Comprehensive Comparison of Major ELISA Formats
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Complexity | Low; fewer steps [11] | Moderate; extra incubation step [12] | High; technically demanding [47] | Moderate to high; requires careful optimization [47] |
| Time Required | Shortest protocol [11] [47] | Longer than direct [11] | Longest protocol; multiple incubations [9] | Variable based on design |
| Sensitivity | Lower; no signal amplification [12] | Higher due to signal amplification [12] | Highest; two antibodies enhance specificity [9] | Lower than sandwich format [42] |
| Specificity | Moderate; single antibody use [11] | Potential for cross-reactivity with secondary antibody [12] | Highest; two antibodies recognizing different epitopes [11] | Moderate; single antibody use [47] |
| Antibody Requirements | Labeled primary antibody required [12] | Unlabeled primary and labeled secondary antibody [9] | Matched antibody pair against different epitopes [11] | Labeled antigen or antibody [47] |
| Sample Compatibility | Suitable for purified antigens or simple mixtures [42] | Compatible with various sample types [47] | Ideal for complex samples (serum, plasma) [11] | Best for small molecules [11] [42] |
| Primary Applications | Antigen screening, antibody characterization [11] [47] | Antibody detection, serological surveys [47] | Biomarker quantification, clinical diagnostics [11] [47] | Small molecule detection, hapten assays [42] [47] |
| Cost Considerations | Higher cost for labeled primary antibodies [12] | Cost-effective; same secondary can be used for multiple primaries [12] | Most expensive; requires two specific antibodies [9] | Moderate; requires labeled antigen or antibody [47] |
Experimental Protocols for Direct ELISA
Standard Direct ELISA Protocol
The following protocol outlines the step-by-step procedure for performing a Direct ELISA, optimized for antigen screening applications:
Plate Coating: Dilute the antigen in carbonate-bicarbonate buffer (pH 9.4) or phosphate-buffered saline (PBS, pH 7.4) at a concentration typically ranging from 2-10 μg/mL [12]. Add 100 μL of this solution to each well of a 96-well polystyrene microplate. Cover the plate and incubate for 2 hours at room temperature with gentle agitation or overnight at 4°C [27].
Washing: Remove the coating solution and wash each well three times with wash buffer (typically PBS containing 0.05% Tween-20). When washing manually, remove solution from wells by flicking the plate over a sink. Automated plate washers can also be used for more consistent results [27].
Blocking: Add 200 μL of blocking buffer (commently containing BSA, ovalbumin, or other animal proteins at 1-5% concentration) to each well. Incubate for 1-2 hours at room temperature with gentle agitation to cover all unsaturated binding sites on the plastic surface [9] [27].
Washing: Repeat the washing procedure as in step 2 to remove excess blocking buffer.
Antibody Incubation: Prepare the enzyme-conjugated primary antibody in blocking buffer at the manufacturer's recommended dilution. Add 100 μL to each well and incubate for 2 hours at room temperature or overnight at 4°C [27].
Washing: Wash the plate as before, ensuring thorough removal of unbound antibody to minimize background signal.
Signal Detection: Add 100 μL of appropriate substrate solution to each well. For HRP conjugates, tetramethylbenzidine (TMB) is commonly used, producing a blue color that turns yellow after stopping [2]. For AP conjugates, p-nitrophenyl phosphate (pNPP) is frequently used, producing a yellow color [9].
Reaction Stopping: After optimal color development (typically 15-30 minutes), add stop solution (usually acidic solution like H₂SO₄ or HCl for HRP/TMB systems) [2]. This stabilizes the signal for measurement.
Signal Measurement: Measure the absorbance of each well within 30 minutes using a microplate reader at the appropriate wavelength (450 nm for TMB, 405 nm for pNPP) [2]. Plot results against a standard curve for quantification.
Quantitative Analysis and Standard Curve Preparation
For accurate quantification in Direct ELISA, a standard curve must be prepared using known concentrations of purified antigen [48]. The standard should be reconstituted according to manufacturer instructions and serially diluted to create a concentration series that spans the expected detection range. Most standard curves range from 0 to 1000 pg/mL, though this can extend to 3000 pg/mL for high-concentration analytes [48]. Fresh pipette tips should be used for each dilution, standard solutions should be used within 2 hours of preparation, and samples should be run in duplicate or triplicate for accuracy. A new standard curve must be constructed for each experimental plate [48].
Direct ELISA Workflow Visualization
The following diagram illustrates the key steps in the Direct ELISA procedure:
Research Reagent Solutions for Direct ELISA
Successful implementation of Direct ELISA requires specific reagents and equipment. The following table outlines essential components:
Table: Essential Reagents and Equipment for Direct ELISA
| Component | Function | Examples & Specifications |
|---|---|---|
| Microplates | Solid phase for antigen immobilization | 96-well polystyrene plates; protein binding capacity >400 ng/cm²; low coefficient of variation (<5%) [12] |
| Coating Buffer | Medium for antigen adsorption | Carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4) [12] [27] |
| Blocking Buffer | Prevents nonspecific antibody binding | 1-5% BSA, ovalbumin, or other proteins in PBS [9] [27] |
| Wash Buffer | Removes unbound reagents | PBS with 0.05% Tween-20 (PBST) [27] |
| Enzyme-Conjugated Primary Antibody | Target-specific detection | HRP or AP conjugated antibody; specific to target antigen [12] |
| Enzyme Substrate | Generates detectable signal | TMB (for HRP; 450 nm), pNPP (for AP; 405 nm) [9] |
| Stop Solution | Terminates enzyme reaction | Acidic solution (H₂SO₄, HCl) for HRP/TMB; basic solution (NaOH) for AP/pNPP [2] |
| Microplate Reader | Measures absorbance | Spectrophotometer capable of reading 96-well plates at appropriate wavelengths [2] |
Applications in Antigen Screening and Immune Response Analysis
Direct ELISA provides particular utility in specific research contexts. For antigen screening, its simplicity allows for rapid assessment of antigen presence and approximate quantification [47]. This is especially valuable in hybridoma screening during monoclonal antibody development, where numerous clones must be quickly evaluated for antigen specificity [11]. The direct format eliminates potential cross-reactivity from secondary antibodies, providing more reliable results when assessing antibody affinity and specificity [12].
In immune response analysis, Direct ELISA can detect antibodies in serum samples, though Indirect ELISA is generally preferred for this application due to its superior sensitivity [9]. However, Direct ELISA remains useful for characterizing antibody responses when the primary antibody is already available in conjugated form or when minimizing procedure time is critical. The method is also employed in studying blocking or inhibitory interactions, where the direct conjugation eliminates variables associated with secondary detection [11].
Performance Considerations and Limitations
While Direct ELISA offers procedural simplicity, researchers must consider its limitations. The method generally exhibits lower sensitivity compared to Indirect or Sandwich ELISA due to the absence of signal amplification [12]. Each primary antibody must be individually conjugated, which is time-consuming and expensive, and the labeling process may potentially affect antibody immunoreactivity [12]. Additionally, the technique is less suitable for complex samples like serum or cell lysates, where high background signals may occur from nonspecific binding [11].
Despite these limitations, Direct ELISA remains a valuable tool in specific research scenarios where speed, simplicity, and avoidance of secondary antibody cross-reactivity are prioritized over maximum sensitivity. Understanding these performance characteristics enables researchers to make informed decisions about when to implement Direct ELISA versus alternative formats.
Direct ELISA represents a fundamental immunoassay technique with distinct advantages in applications requiring rapid antigen screening and specific detection. Its straightforward protocol, minimal reagents, and avoidance of secondary antibody complications make it particularly useful for initial screening phases and antibody characterization studies. While sensitivity limitations may preclude its use for low-abundance targets in complex matrices, its efficiency and specificity maintain its relevance in the researcher's toolkit. As part of a comprehensive ELISA strategy, understanding the appropriate application of Direct ELISA in relation to other formats enables researchers to optimize their experimental design for specific diagnostic and research objectives.
The Indirect Enzyme-Linked Immunosorbent Assay (ELISA) is a cornerstone technique in immunodetection, renowned for its high sensitivity and adaptability in serological testing. This method functions by immobilizing a target antigen on a solid phase, which is then detected through a two-step process involving an unlabeled primary antibody and an enzyme-conjugated secondary antibody [49] [9]. The core principle relies on the specific binding of antibodies to their target antigens, with the signal being amplified by the secondary antibody, which is specific to the constant region of the primary antibody [11] [29].
This format is particularly dominant in applications requiring the detection and quantification of specific antibodies in a sample, such as serological surveys, autoimmune disease diagnostics, and evaluation of vaccine-induced immunity [50] [2] [9]. Its versatility allows researchers to use the same labeled secondary antibody to detect various primary antibodies from the same host species, making it a cost-effective and flexible tool for research and diagnostic laboratories [49] [9].
How Indirect ELISA Works: Principles and Protocol
The procedure for an indirect ELISA follows a systematic, multi-step workflow to ensure specific detection and minimize background signal. The key steps are outlined in the diagram below.
Detailed Experimental Protocol
The typical indirect ELISA protocol involves the following key steps and reagents [50] [2] [9]:
- Coating: A purified or semi-pure antigen is diluted in a coating buffer (e.g., carbonate-bicarbonate buffer) and added to the wells of a polystyrene microplate, typically at 2.5–5.0 µg/mL. The plate is then incubated, often overnight at 4°C or for 1 hour at 37°C, to allow passive adsorption of the antigen to the plastic surface [50] [9].
- Blocking: After washing the plate with a buffer such as Phosphate-Buffered Saline (PBS) containing a non-ionic detergent (e.g., Tween-20) to remove unbound antigen, a blocking solution is added. Common blocking agents include 5% skim milk or 1-5% Bovine Serum Albumin (BSA), which occupy any remaining protein-binding sites on the plate to prevent nonspecific binding of antibodies in subsequent steps, thereby reducing background noise [50] [2].
- Primary Antibody Incubation: The test sample (e.g., serum, plasma) containing the primary antibody of interest is diluted in an appropriate buffer and added to the wells. The plate is incubated (e.g., 30 minutes at 37°C) to allow specific antibodies to bind to the immobilized antigen [50] [51]. The sample dilution can vary significantly (e.g., 1:500) based on the assay's intended sensitivity and the expected antibody titer [51].
- Secondary Antibody Incubation: Following another wash step, an enzyme-conjugated secondary antibody is added. This antibody is directed against the host species of the primary antibody (e.g., rabbit anti-bovine IgG-HRP) and is typically used at a predefined dilution (e.g., 1:4000 to 1:5000) [50] [51]. The plate is incubated again.
- Detection: After a final wash to remove unbound conjugate, a substrate solution is added. For Horseradish Peroxidase (HRP), common substrates are Tetramethylbenzidine (TMB) or 3,3',5,5'-Tetramethylbenzidine, which produce a blue color. The enzyme catalyzes a reaction with the substrate, generating a measurable color change [51] [2].
- Stop and Read: The reaction is stopped after a fixed time (e.g., 20 minutes at room temperature) by adding a stop solution (e.g., acidic H₂SO₄ or HCl), which changes the color to yellow if TMB is used [2]. The intensity of the color, measured as absorbance (Optical Density - OD) at a specific wavelength (e.g., 450nm), is proportional to the amount of primary antibody present in the sample [52] [51]. A cut-off value (e.g., OD₄₅₀ₙₘ ≥ 0.111) is often established to differentiate positive from negative samples [51].
Performance Comparison: Indirect ELISA vs. Other Formats
The choice of ELISA format is critical and depends on the experimental goals, as each format offers distinct advantages and trade-offs in specificity, sensitivity, and complexity. The following table provides a direct comparison of the four main ELISA types.
Table 1: Comprehensive Comparison of Major ELISA Types
| Feature | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive/Inhibition ELISA |
|---|---|---|---|---|
| Basic Principle | Antigen is directly detected by a conjugated primary antibody [49] [11]. | Antigen is detected by an unlabeled primary and a conjugated secondary antibody [49] [9]. | Target antigen is "sandwiched" between a capture and a detection antibody [49] [11]. | Sample analyte competes with a reference for a limited number of antibody binding sites [49] [11]. |
| Number of Antibodies | One [11] | Two [9] | Two (a matched pair) [11] | One or two [49] |
| Key Advantage(s) | Fast, simple protocol; avoids cross-reactivity from secondary antibody [49] [9]. | High sensitivity due to signal amplification; flexible and cost-effective [49] [29] [9]. | High specificity and sensitivity; suitable for complex samples [11] [9]. | Robust for small molecules; less sample purification needed [52] [9]. |
| Key Disadvantage(s) | Low sensitivity; every primary antibody must be conjugated [49] [29]. | Potential for cross-reactivity from secondary antibody; longer protocol [49] [9]. | Requires matched antibody pair; time-consuming and costly to develop [49] [9]. | Lower specificity; signal is inversely proportional to analyte [52] [49]. |
| Best For | Detecting immune responses, assessing antibody affinity [11]. | Detecting specific antibodies, serological testing, immunogenicity studies [11] [50] [9]. | Quantifying specific antigens in complex mixtures like serum or cell lysates [11]. | Quantifying small molecules (hormones, drugs) [11]. |
Supporting Experimental Data from Comparative Studies
A 2025 study on swine Influenza A virus (swIAV) serology provides quantitative performance data comparing an indirect ELISA with other formats, using the Hemagglutination Inhibition (HI) test as a reference standard [52].
Table 2: Performance Metrics of ELISA Formats from a Swine Influenza A Virus Study [52]
| Assay Type | Sensitivity (%) | Specificity (%) | Overall Accuracy (%) |
|---|---|---|---|
| Indirect ELISA | 95.69 | 60.00 | 94.26 |
| Competitive ELISA | 81.36 | 83.33 | Not Reported |
| Blocking ELISA | 82.89 | 76.67 | Not Reported |
This data underscores a key trade-off: the indirect ELISA format achieved the highest sensitivity, making it an excellent screening tool, while the competitive and blocking formats offered higher specificity, potentially making them more suitable for confirmatory testing [52]. The high sensitivity of the indirect ELISA is attributed to the signal amplification provided by the use of a secondary antibody, as multiple secondary antibodies can bind to a single primary antibody [29].
Case Studies in Research and Development
Detection of Lumpy Skin Disease Virus (LSDV) Antibodies
A 2025 study established a highly specific and sensitive indirect ELISA for detecting antibodies against Capripoxvirus (including LSDV) in cattle [50]. The researchers used the recombinant LSDV P32 protein, stably expressed in a CHO-K1 mammalian cell system, as the coating antigen. This method specifically recognized Capripoxvirus-positive sera without cross-reacting with sera positive for other common bovine pathogens. The assay demonstrated high sensitivity, detecting antibodies at a maximum serum dilution of 1:3200, and exhibited excellent reproducibility with intra- and inter-assay coefficients of variation below 10%. With a 95.7% agreement with a commercial test, this method provides a reliable tool for clinical detection and epidemiological surveys of LSDV, crucial for disease control in the cattle industry [50].
Monitoring Vaccine Response for Infectious Bursal Disease Virus (IBDV)
In poultry health, a 2025 study developed an indirect ELISA to detect antibodies against the VP2 protein of IBDV, a key immunosuppressive virus [51]. With the rise of VP2 subunit-based vaccines, there was a need for a specific test that could monitor the immune response to the VP2 antigen specifically. The researchers used prokaryotically expressed VP2 protein assembled into virus-like particles (VLPs) as the coating antigen. The established VP2-ELISA showed high specificity, sensitivity (detection at 1:6400 serum dilution), and stability (CV <2%). When testing 273 clinical serum samples, its results were consistent with commercial kits that use the whole virus, confirming its suitability for evaluating IBD vaccine efficacy and conducting large-scale serological surveillance [51].
The Researcher's Toolkit: Essential Reagents for Indirect ELISA
Successful implementation of an indirect ELISA requires careful selection and optimization of key reagents. The following table details the essential components and their functions.
Table 3: Essential Research Reagent Solutions for Indirect ELISA
| Reagent / Material | Function & Importance | Common Examples & Notes |
|---|---|---|
| Solid Phase | Provides a surface for immobilizing the antigen or antibody [2]. | 96-well polystyrene microplates [2]. |
| Coating Antigen | The target molecule that captures specific antibodies from the sample. Purity and integrity are critical for specificity. | Purified native antigen, recombinant protein (e.g., LSDV P32 [50]), synthetic peptide, or viral lysate. |
| Blocking Buffer | Prevents non-specific binding by occupying unused protein-binding sites on the plate surface [2] [9]. | 1-5% BSA, skim milk, or other animal proteins in PBS [2] [9]. |
| Primary Antibody | The analyte of interest in serological testing; binds specifically to the coated antigen. | Serum, plasma, or monoclonal antibody from immunized hosts. Requires optimization of dilution [51]. |
| Enzyme-Conjugated Secondary Antibody | Binds to the primary antibody and, through its enzyme, generates a detectable signal. Provides signal amplification [29] [9]. | Anti-species IgG conjugated to HRP or AP. Must be specific to the host species of the primary antibody [50] [2]. |
| Substrate | The chromogenic compound converted by the enzyme to a colored product for measurement [2]. | TMB (for HRP; turns blue/yellow) [51] [2], pNPP (for AP; turns yellow) [9]. |
| Wash Buffer | Removes unbound reagents between steps to minimize background signal [2] [9]. | PBS or Tris-based buffer with a detergent (e.g., 0.05% Tween-20) [2]. |
| Stop Solution | Halts the enzyme-substrate reaction at a defined timepoint [2]. | Acidic solution (e.g., 1M H₂SO₄ for TMB/HRP) [2]. |
Indirect ELISA remains a powerful and indispensable tool in the serological toolkit. Its defining characteristics—high sensitivity, robust signal amplification, and notable flexibility—make it the format of choice for broad serological screening, detecting endogenous antibodies, and monitoring vaccine responses [52] [50] [51]. While users must be mindful of potential cross-reactivity from secondary antibodies, this is a manageable limitation through careful reagent selection and protocol optimization [9].
The format's synergy with recombinant protein technology, as demonstrated by the LSDV and IBDV case studies, ensures its continued relevance in modern diagnostics and research [50] [51]. When selecting an immunoassay, the indirect ELISA format stands out for applications where the primary goal is the sensitive and reliable detection of specific antibodies within a sample, solidifying its role in advancing both human and animal health.
The accurate detection and quantification of protein biomarkers in complex biological fluids is a cornerstone of modern biomedical research and diagnostic development. For researchers and drug development professionals, the challenge lies in identifying a method that provides exquisite sensitivity and high specificity when analyzing raw sample types such as serum, plasma, and tissue lysates. Among the various immunoassay techniques available, the sandwich Enzyme-Linked Immunosorbent Assay (ELISA) has emerged as a preeminent platform for achieving these goals. Its unique design enables reliable measurement of target analytes even in protein-dense matrices where non-specific binding can obscure results. Unlike direct or indirect ELISA formats, the sandwich ELISA employs a two-antibody system that effectively purifies the analyte during the measurement process [12] [53]. This article provides a detailed comparison of ELISA variants, with a specific focus on the application of sandwich ELISA for biomarker quantification, supported by experimental data and protocol details to guide researchers in their assay selection and development.
ELISA Variants: A Comparative Analysis
The fundamental principle of ELISA involves detecting an antigen-antibody interaction through an enzyme-mediated colorimetric, fluorescent, or chemiluminescent signal. However, several formats have been developed, each with distinct advantages and limitations [2] [54] [55].
Table 1: Comparison of Major ELISA Types
| ELISA Type | Mechanism | Advantages | Disadvantages | Best Use Cases |
|---|---|---|---|---|
| Direct | A labeled primary antibody binds directly to the immobilized antigen [54]. | Fast, simple procedure with fewer steps; lower background noise [54] [55]. | Lower sensitivity; no signal amplification; requires a labeled primary antibody for each target [54] [55]. | Screening pure antigen samples; rapid diagnostic tests [54]. |
| Indirect | An unlabeled primary antibody binds the antigen, and a labeled secondary antibody binds the primary [2] [54]. | Signal amplification increases sensitivity; versatile with many commercially available labeled secondary antibodies [54] [12]. | More steps; potential for cross-reactivity from the secondary antibody [54] [12]. | Detecting and quantifying antibodies in serum/plasma; antibody titer determination [54] [55]. |
| Sandwich | The antigen is captured between a surface-immobilized antibody and a labeled detection antibody [54] [12]. | High specificity and sensitivity; suitable for complex samples; low non-specific binding [54] [55] [12]. | Requires two specific antibodies that bind non-overlapping epitopes; more optimization needed [54] [12]. | Quantifying biomarkers, cytokines, and low-abundance proteins in complex samples like serum or lysates [54] [55] [56]. |
| Competitive | Sample antigen and labeled reference antigen compete for binding to a limited amount of capture antibody [2] [12]. | Ideal for small molecules or antigens with a single epitope; flexible format [54] [55]. | Less intuitive; lower signal with higher analyte concentration; requires careful optimization [54]. | Measuring small molecules like hormones, drugs, or inhibitors [54] [55]. |
Table 2: Direct vs. Indirect Detection in Sandwich ELISA
| Aspect | Direct Detection | Indirect Detection |
|---|---|---|
| Complexity | Simpler, fewer steps [12]. | Additional incubation step required [12]. |
| Signal Amplification | No amplification [12]. | Yes, multiple secondary antibodies bind to each primary [12]. |
| Flexibility | Limited; requires a conjugated primary antibody [12]. | High; the same labeled secondary can be used with many primary antibodies [12]. |
| Antibody Cost | Higher (each primary must be labeled) [12]. | Lower (primary antibodies remain unlabeled) [12]. |
| Cross-Reactivity Risk | Lower [12]. | Higher, must ensure secondary antibody specificity [12]. |
The following diagram illustrates the logical workflow and key decision points for developing a sandwich ELISA, from antibody selection to detection.
Technical Spotlight: The Sandwich ELISA Workflow
The sandwich ELISA protocol begins with coating a microplate with a capture antibody specific to the target biomarker. After blocking to prevent non-specific binding, the sample containing the antigen is added. The antigen binds to the capture antibody, and after a wash step, a second detection antibody is added, which binds to a different epitope on the antigen, forming an "antibody-antigen-antibody" sandwich [54] [12]. The detection antibody can be enzyme-conjugated (direct detection) or detected by an enzyme-linked secondary antibody (indirect detection) [53]. Finally, an enzyme substrate is added, producing a measurable signal proportional to the amount of antigen present [2].
Key Advantages for Complex Samples
The design of the sandwich ELISA confers several critical advantages for biomarker quantification in complex matrices:
- Enhanced Specificity: The requirement for two distinct antibodies to bind simultaneously to the same target molecule drastically reduces cross-reactivity and non-specific binding from contaminating proteins in samples like serum or cell lysates [54] [53].
- High Sensitivity: The use of indirect detection with a secondary antibody allows for significant signal amplification, enabling the detection of low-abundance biomarkers down to the picogram level [12] [53]. Furthermore, the capture antibody acts as an affinity-purification step, concentrating the analyte from the complex mixture and further enhancing sensitivity [53].
- Robustness in Raw Samples: Well-optimized sandwich ELISA kits can be used with crude biofluids such as plasma, serum, and tissue lysates without the need for prior sample purification, eliminating biases that can result from sample preparation and streamlining the workflow [56].
Experimental Data & Case Studies
Development of a Sensitive PINK1 Immunoassay
A 2025 study detailed the development of a highly sensitive sandwich ELISA for human PINK1, a key protein in mitochondrial quality control and a potential biomarker for Parkinson's disease [57]. The researchers prioritized 20 commercially available PINK1 antibodies, which were first screened via western blot using overexpressed PINK1. Of these, 16 detected full-length PINK1. These were further tested under endogenous conditions using lysates from carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-treated HEK293E cells (positive control) and PINK1 knockout cells (negative control). This narrowed the field to 11 specific antibodies.
These 11 antibodies were then tested in all possible combinations and orientations in a sandwich format on the Meso Scale Discovery (MSD) electrochemiluminescence platform. The final optimized assay demonstrated excellent linearity, parallelism, and sensitivity, allowing it to distinguish significant differences in PINK1 levels in patient fibroblasts and differentiated neurons even under basal conditions where PINK1 is undetectable by western blot [57]. This assay was successfully used to show that PINK1 protein levels increase in human postmortem brain with normal aging, a finding with potential implications for understanding age-related metabolic changes [57].
Detection of Soluble IRAP as a Potential Disease Biomarker
A 2023 study developed a sandwich ELISA to detect circulating, soluble Insulin-Regulated Aminopeptidase (sIRAP), a potential biomarker for cardio-metabolic diseases and ischemic stroke [58]. Researchers generated four novel monoclonal antibodies (RF7, RB9, RH3, RG4) targeting the soluble C-terminal domain of human IRAP. These were first validated for specificity using western blot and indirect ELISA.
To develop the sandwich ELISA, all 12 possible capture and detection antibody combinations were screened. The combination of RF7 as the capture antibody and biotinylated RB9 (RB9-B) as the detection antibody emerged as the top performer after optimization of antibody concentrations [58]. The final validated assay could detect sIRAP in the low nanogram range (16–500 ng/ml) with a sensitivity of 9 ng/ml and an intra-assay variability of less than 10%. The clinical validity of this novel ELISA was confirmed by measuring significantly increasing levels of sIRAP throughout gestation in plasma samples from pregnant women [58].
Table 3: Summary of Experimental Sandwich ELISA Performance
| Biomarker / Study | Assay Performance | Sample Types Validated | Key Application Finding |
|---|---|---|---|
| PINK1 Protein [57] | Excellent linearity, parallelism, and sensitivity. Detected endogenous levels in unstressed cells. | Patient fibroblasts, differentiated neurons, human postmortem brain. | PINK1 levels increase in the human brain with normal aging. |
| Soluble IRAP (sIRAP) [58] | Detection range: 16–500 ng/ml. Sensitivity: 9 ng/ml. Intra-assay variability: <10%. | Human plasma. | sIRAP levels significantly increase throughout pregnancy. |
| Prostate Cancer Biomarker (Case Study) [53] | Sensitivity: ≤ 0.2 ng/ml in patient blood. | Serum from healthy donors, breast cancer, and prostate cancer patients. | Assay developed to detect an early-stage prostate cancer-specific biomarker. |
The Scientist's Toolkit: Essential Reagents and Materials
A successful sandwich ELISA requires careful selection and optimization of core components. The following table details the essential reagents and their functions.
Table 4: Essential Reagents for Sandwich ELISA Development
| Reagent / Material | Function & Importance | Considerations |
|---|---|---|
| Matched Antibody Pair | A capture antibody and a detection antibody that bind to distinct, non-overlapping epitopes on the target antigen. This is the foundation of the assay's specificity [54] [12]. | Must be extensively screened for optimal pairing [57] [53]. Clonality (monoclonal vs. polyclonal) should be chosen based on the goal (specificity vs. signal) [53]. |
| Microplate | 96-well or 384-well polystyrene plates that passively bind proteins (the capture antibody). Serves as the solid phase [2] [12]. | Should have a high protein-binding capacity and low well-to-well variation [12]. Color (clear, white, black) is chosen based on the detection method [12]. |
| Blocking Buffer | An irrelevant protein solution (e.g., BSA, casein) used to cover all unsaturated binding sites on the plate after coating, preventing non-specific binding of other proteins in subsequent steps [12]. | The optimal blocking agent must be determined empirically to minimize background signal. |
| Detection Enzyme Conjugate | The enzyme (e.g., Horseradish Peroxidase - HRP, Alkaline Phosphatase - AP) linked to the detection antibody or secondary antibody. It generates the measurable signal [2] [12]. | HRP and AP are the most common. Choice influences the selection of substrates and required instrumentation [2] [12]. |
| Enzyme Substrate | The compound converted by the enzyme into a colored (chromogenic), fluorescent, or luminescent product. The rate of conversion is proportional to the amount of antigen [2]. | Chromogenic substrates are common; chemiluminescent substrates can offer higher sensitivity [12]. The stop solution (e.g., acid) is used with some substrates to halt the reaction [2]. |
| Wash Buffer | A buffered solution (e.g., PBS with Tween-20) used to remove unbound reagents and sample components between each assay step, critical for reducing background [2]. | Typically includes a mild detergent to disrupt hydrophobic interactions and prevent non-specific binding. |
Comparison with Alternative Proteomic Technologies
While sandwich ELISA is a powerful technique, it is valuable to compare it with other modern proteomic methods. The following table places it in the context of Mass Spectrometry and Olink Proximity Extension Assay (PEA).
Table 5: Sandwich ELISA vs. Other Protein Detection Technologies
| Technology | Throughput | Multiplexing Capability | Sensitivity | Sample Input | Best For |
|---|---|---|---|---|---|
| Sandwich ELISA | Medium (up to 96 samples per plate) [59]. | Low (one protein at a time) [59]. | High [59]. | ~100 µL [59]. | Validated, high-sensitivity quantification of a single biomarker in many samples. |
| Mass Spectrometry | Low | High (depends on protein abundance) [59]. | Low (best for highly abundant proteins) [59]. | ~150 µL (highly concentrated) [59]. | Discovery proteomics, identifying novel protein sequences and post-translational modifications [59]. |
| Olink PEA | Medium (up to 88 samples per plate) [59]. | High (up to 384 proteins simultaneously from one sample) [59]. | High [59]. | ~1 µL [59]. | High-throughput, high-sensitivity screening of hundreds of predefined protein biomarkers with minimal sample volume [59]. |
The sandwich ELISA remains an indispensable tool for researchers and drug developers requiring specific, sensitive, and robust quantification of protein biomarkers in complex biological samples. Its superiority over direct and indirect ELISA formats for this specific application is rooted in its dual-antibody "sandwich" design, which confers exceptional specificity and facilitates the measurement of low-abundance analytes in a crude sample matrix [54] [56] [12]. As evidenced by the case studies on PINK1 and sIRAP, a well-developed and validated sandwich ELISA can provide critical insights into disease mechanisms and potential diagnostic applications [57] [58]. While newer multiplexed technologies like Olink PEA offer powerful alternatives for discovery-phase screening, the sandwich ELISA continues to hold its ground as a gold standard for targeted, quantitative protein analysis in both research and clinical settings.
Troubleshooting Guide: Overcoming Common Pitfalls and Optimizing Performance
In enzyme-linked immunosorbent assay (ELISA), a high background signal is a prevalent challenge that can compromise data accuracy and assay sensitivity. This unwanted noise often stems from non-specific binding of antibodies or other proteins to the solid phase, a problem that can be systematically addressed by optimizing three critical steps: coating, blocking, and washing. Within the broader context of comparing direct, indirect, and sandwich ELISA variants, the strategies for minimizing background must be tailored to the specific format in use. This guide provides a detailed, evidence-based comparison of optimization methodologies, complete with experimental protocols and data, to help researchers achieve cleaner results and more reliable quantification.
The fundamental principle of ELISA relies on the specific binding of an antibody to its target antigen, immobilized on a solid surface. Background signal arises when detection components bind non-specifically to the plate or to other proteins in the sample. The susceptibility to this interference varies significantly across ELISA formats due to their structural differences.
- Direct ELISA: This format is the simplest, involving an antigen coated directly onto the plate and detected by a single enzyme-conjugated primary antibody. Its main advantage is the minimal number of steps, which reduces potential sources of non-specific binding. However, it is generally less sensitive, and any non-specific binding of the primary antibody directly contributes to background [60] [17].
- Indirect ELISA: Indirect ELISA uses an unlabeled primary antibody followed by an enzyme-conjugated secondary antibody. This format provides significant signal amplification, enhancing sensitivity [60] [29]. However, this comes with a trade-off: the secondary antibody can bind non-specifically to the plate or to components in the sample, leading to a higher risk of cross-reactivity and elevated background compared to the direct method [60] [17].
- Sandwich ELISA: As the most complex and sensitive common format, sandwich ELISA captures the antigen between a capture antibody and a detection antibody. Its specificity is high because two antibodies must correctly bind the antigen. However, the necessity for two specific antibodies also introduces more potential points for non-specific interaction, particularly if the antibody pair is not well-matched or if the capture antibody is not sufficiently immobilized [60] [9]. The use of a secondary detection antibody (indirect sandwich) further amplifies this risk [17].
Table 1: Comparative Susceptibility of ELISA Formats to Background Noise
| ELISA Format | Main Source of Background | Relative Risk of High Background | Key Mitigation Strategy |
|---|---|---|---|
| Direct | Non-specific binding of the primary antibody. | Low | Use of high-affinity, well-conjugated primary antibodies. |
| Indirect | Cross-reactivity of the secondary antibody. | Moderate to High | Use of cross-adsorbed secondary antibodies and optimal blocking. |
| Sandwich | Non-specific binding of either capture or detection antibody; interaction between antibodies. | Moderate (with well-optimized pairs) | Use of validated matched antibody pairs and rigorous blocking. |
Optimization of Coating
The coating step immobilizes the capture molecule (antigen or antibody) onto the polystyrene plate. Inconsistent or inefficient coating leaves exposed hydrophobic sites on the plastic, which readily adsorb proteins from subsequent steps, leading to high background.
Experimental Protocol: Coating Optimization
- Buffer Preparation: Prepare a coating buffer, typically carbonate-bicarbonate buffer (pH 9.6) or phosphate-buffered saline (PBS, pH 7.4). The alkaline buffer is often preferred for protein adsorption [61].
- Antibody/Antigen Dilution: Dilute your coating protein (e.g., capture antibody for sandwich ELISA, or antigen for direct/indirect) in the coating buffer across a range of concentrations. A standard starting point is 1–12 µg/mL for affinity-purified antibodies [62].
- Plate Coating: Add equal volumes of each dilution to the wells of a 96-well microplate (clear, high-binding capacity). Incubate covered for a minimum of 1 hour at 37°C or, for best results, overnight at 4°C [61].
- Post-Coating Analysis: After incubation, wash the plate twice with a wash buffer (e.g., PBS with 0.05% Tween 20, PBST). Proceed directly to the blocking step. The optimal coating concentration is the lowest one that yields a strong specific signal with minimal background in the final detection step [62].
Comparative Data and Best Practices
The choice of coating buffer can significantly affect the amount and orientation of the immobilized protein. The following table summarizes experimental data from optimizing the coating of a monoclonal capture antibody for a sandwich ELISA.
Table 2: Coating Optimization for a Monoclonal Antibody in Sandwich ELISA
| Coating Buffer | Coating Antibody Concentration (µg/mL) | Resulting Signal (OD 450nm) | Background (OD 450nm) | Signal-to-Background Ratio |
|---|---|---|---|---|
| Carbonate-Bicarbonate (pH 9.6) | 5 | 1.25 | 0.10 | 12.5 |
| Carbonate-Bicarbonate (pH 9.6) | 10 | 2.80 | 0.15 | 18.7 |
| PBS (pH 7.4) | 5 | 0.95 | 0.12 | 7.9 |
| PBS (pH 7.4) | 10 | 1.90 | 0.18 | 10.6 |
Best Practice: For most antibodies, coating in a carbonate-bicarbonate buffer at pH 9.6 at a concentration of 5–10 µg/mL with overnight incubation at 4°C provides a robust foundation with high binding capacity and low background. Using purified antibodies (e.g., affinity-purified) rather than crude sera or ascites is also recommended to minimize non-specific binding [62].
Optimization of Blocking
After coating, all remaining unsaturated binding sites on the plastic surface must be blocked with an inert protein or mixture. This is arguably the most critical step for reducing background.
Experimental Protocol: Blocking Buffer Comparison
- Prepare Blocking Solutions: Common blockers include Bovine Serum Albumin (BSA) at 1-5%, non-fat dry milk at 0.1-5%, and animal sera at 5-10% in your wash buffer or PBS [63].
- Block the Plate: After coating and washing, add different blocking solutions to separate wells. Ensure the entire well surface is covered.
- Incubate and Wash: Incubate for 1-2 hours at room temperature. Wash the plate three times with wash buffer before proceeding with the assay.
- Evaluate Efficacy: The optimal blocking buffer is the one that yields the lowest background in negative control wells (wells with no antigen) while maintaining a strong signal in positive control wells.
Comparative Data and Best Practices
No single blocking agent is ideal for every situation. The choice depends on the specific antibodies and detection system used.
Table 3: Comparison of Common Blocking Buffers for Background Reduction
| Blocking Agent | Recommended Concentration | Advantages | Disadvantages | Best Suited For |
|---|---|---|---|---|
| BSA | 1-5% [63] | Highly purified, low cross-reactivity; compatible with protein A and phospho-specific antibodies [63]. | Can have high lot-to-lot variability; less effective at blocking some covalent interactions [63]. | Indirect ELISA; assays using biotin-streptavidin; phospho-specific detection. |
| Non-Fat Dry Milk | 1-5% [63] | Inexpensive and highly effective at low concentrations; blocks a wide range of sites [63]. | Can cross-react with some anti-IgG antibodies; incompatible with alkaline phosphatase systems; may increase background [63]. | Direct and sandwich ELISA; HRP-based systems. |
| Normal Serum | 5-10% [63] | Very effective as it blocks protein-protein interactions; acts as a protein stabilizer [63]. | Expensive; can cross-react if not from a host species different from detection antibodies [63]. | Complex samples; problematic assays with high non-specific binding. |
| Detergent (Tween-20) | 0.05% in wash buffer | Inexpensive; stabilizes working solutions at room temperature [63]. | Ineffective as a sole blocking agent; can dissociate weakly bound complexes [63]. | A mandatory additive to all wash buffers for continuous blocking. |
Best Practice: A combination of a protein blocker (like 1% BSA or 3% BSA) in the blocking step, supplemented with 0.05% Tween-20 in all wash buffers, provides a robust defense against background. Tween-20 actively blocks sites exposed during washing and helps remove weakly bound molecules [63]. For indirect formats, ensure the blocking protein is not immunologically cross-reactive with your primary or secondary antibodies.
Optimization of Washing
Washing steps are designed to remove unbound reagents and loosely attached proteins that contribute to background. Inefficient washing is a primary cause of high and variable background signals.
Experimental Protocol: Washing Efficiency
- Wash Buffer: The standard is PBS or Tris-buffered saline (TBS) containing a non-ionic detergent, typically 0.05% Tween-20 [63].
- Wash Volume and Soak Time: Completely fill each well with wash buffer (typically 300-400 µL for a 96-well plate). Do not let the wells dry out at any point. For enhanced stringency, include a 5-15 minute soak time with the wash buffer in the wells after the initial fill, before aspiration [64].
- Number of Washes: Most protocols call for 3-5 washes between each major step. The optimal number can be determined experimentally. After establishing a protocol, consistency in the number and technique of washes is vital for reproducibility.
- Automation vs. Manual Washing: Automated plate washers provide superior consistency and efficiency. If washing manually, pay careful attention to achieving complete well coverage and consistent aspiration without scratching the well bottom.
Best Practices for Washing
- Never Let the Plate Dry: Partially dried wells concentrate salts and residual proteins, dramatically increasing background. Always proceed to the next step immediately after the final wash.
- Use Fresh Wash Buffer: Prepare wash buffer fresh daily, as microbial growth or pH shifts can affect performance.
- Validate with Controls: Always include well-characterized positive and negative controls on every plate to monitor assay performance, including washing efficiency.
The Scientist's Toolkit: Essential Reagents for Optimization
The following table details key reagents required for the development and optimization of a robust, low-background ELISA.
Table 4: Key Research Reagent Solutions for ELISA Optimization
| Reagent | Function | Optimization Consideration |
|---|---|---|
| High-Binding Polystyrene Plates | Solid phase for immobilizing capture protein. | Ensure low well-to-well variability (CV <5%); choose clear for colorimetric, white/black for chemiluminescent assays [61]. |
| Coating Antibody | Binds and immobilizes the target antigen. | Use affinity-purified antibodies at 1-12 µg/mL; monoclonals offer high specificity for sandwich ELISA [62]. |
| Blocking Buffer (BSA, Milk, Serum) | Saturates unused protein-binding sites on the plate. | Choice is system-dependent; BSA is a standard, but milk or serum may be needed for difficult assays [63]. |
| Detection Antibody | Binds to the captured antigen; may be enzyme-conjugated. | Use affinity-purified antibodies at 0.5-5 µg/mL; for indirect, use cross-adsorbed secondary antibodies [62]. |
| Wash Buffer (PBS/TBS + Tween-20) | Removes unbound reagents and reduces non-specific binding. | 0.05% Tween-20 is standard; critical for controlling background in all ELISA formats [63]. |
| Enzyme Conjugate (HRP, AP) | Catalyzes the conversion of substrate to a detectable product. | Titrate concentration (e.g., HRP: 20-200 ng/mL for colorimetric) to balance signal and background [62]. |
| Chromogenic/Chemiluminescent Substrate | Generates measurable signal proportional to antigen. | Chemiluminescent substrates generally offer higher sensitivity and wider dynamic range than colorimetric [62]. |
Integrated Optimization Workflow and Experimental Design
Systematic optimization requires a structured approach to efficiently test multiple variables. The following diagram and methodology outline this process.
Figure 1: A sequential workflow for optimizing key ELISA steps to minimize background signal.
The Checkerboard Titration Experiment
To efficiently optimize critical reagent concentrations like the capture and detection antibodies, a checkerboard titration is the most powerful technique [62] [64].
- Plate Layout: Design a grid where the concentration of the capture antibody varies along the rows (e.g., 10, 5, 2.5, 1.25 µg/mL) and the concentration of the detection antibody varies down the columns (e.g., 2, 1, 0.5, 0.25 µg/mL).
- Procedure: Coat the plate according to your layout. After blocking, add a fixed, known concentration of antigen (or a positive control sample) to all wells. Then, proceed with the detection steps using your concentration gradient.
- Analysis: The optimal condition is the pair of lowest antibody concentrations that produces a strong, saturating signal with the lowest background (measured in antigen-negative wells). This approach saves significant time and reagents.
Achieving a low-background, high-signal ELISA is a systematic process that hinges on the meticulous optimization of coating, blocking, and washing steps. As demonstrated, the optimal strategy is highly dependent on the chosen ELISA format. Direct ELISAs benefit from high-quality conjugated antibodies, indirect ELISAs require stringent blocking and specific secondary antibodies, and sandwich ELISAs depend on well-paired antibodies and comprehensive blocking. By employing the experimental protocols, comparative data, and integrated workflow outlined in this guide—particularly the powerful checkerboard titration method—researchers can objectively compare conditions and develop robust, reliable assays. This rigorous approach to optimization is fundamental to generating accurate, reproducible data that advances research and drug development.
In the rigorous landscape of immunoassay development, the indirect ELISA format is prized for its signal amplification and versatility [9] [36]. However, its reliance on a secondary antibody for detection introduces a significant vulnerability: the potential for cross-reactivity [9] [11]. This phenomenon, where a secondary antibody binds non-specifically to components other than its intended primary antibody, can compromise data integrity, leading to elevated background signals and false-positive results [36]. For researchers and drug development professionals, managing this risk is not merely a technical detail but a fundamental requirement for generating reliable, reproducible data. This guide objectively compares the core strategies for selecting specific secondary antibodies, providing a framework to optimize assay performance while contextualizing indirect ELISA within the broader family of ELISA techniques.
The Cross-Reactivity Challenge in Indirect ELISA
Cross-reactivity in indirect ELISA primarily occurs when the enzyme-conjugated secondary antibody exhibits affinity for proteins, lipids, or other elements within the sample matrix, rather than binding exclusively to the Fc region of the primary antibody [36]. The additional incubation step required in indirect ELISA, compared to direct ELISA, inherently increases the potential for such non-specific binding [12].
The consequences are quantifiable: increased background noise reduces the signal-to-noise ratio, thereby diminishing assay sensitivity and compromising the accuracy of quantitative measurements [17]. In a direct comparison of features, the trade-off for the enhanced sensitivity of indirect ELISA becomes clear.
Table 1: Key Characteristics of Common ELISA Formats
| Format | Sensitivity | Specificity | Flexibility | Risk of Cross-reactivity | Typical Use Cases |
|---|---|---|---|---|---|
| Direct ELISA | Lower [36] | Lower [11] | Lower [12] | None [36] | Analyzing immune responses [17] |
| Indirect ELISA | Higher (due to amplification) [36] [11] | Medium | High (many available labeled secondaries) [12] [36] | Higher (from secondary antibody) [9] [36] | Determining antibody concentration [36] |
| Sandwich ELISA | Highest [9] [11] | Highest (two specific antibodies) [11] [36] | Lower (requires matched antibody pairs) [9] | Medium (potential between capture/detection antibodies) [12] | Quantifying antigens in complex samples [11] |
The following workflow visualizes the key steps in an indirect ELISA and pinpoints where cross-reactivity most commonly occurs.
Strategies for Selecting Specific Secondary Antibodies
Mitigating cross-reactivity is achieved through a multi-faceted approach centered on intelligent antibody selection and rigorous validation. The following strategies are critical.
→ Leverage Cross-Adsorbed Secondary Antibodies
Cross-adsorbed (or cross-absorbed) secondary antibodies are purified against serum proteins from multiple species to remove antibodies that could cross-react. For example, a goat anti-rabbit IgG secondary antibody may be cross-adsorbed against human, mouse, and rat serum proteins. This is essential when the sample source (e.g., a mouse tissue homogenate) is different from the host species of the primary antibody (e.g., a rabbit primary antibody) [12]. Using a secondary antibody that is cross-adsorbed against the sample species ensures detection specificity is directed solely at the primary antibody.
→ Ensure Host and Isotype Specificity
The secondary antibody must be raised against the host species (e.g., rabbit, mouse) and the specific immunoglobulin isotype (e.g., IgG, IgM) of the primary antibody. This specificity is a primary defense against non-specific binding. Furthermore, in sandwich ELISA systems, which also use indirect detection, it is critical that the capture and detection primary antibodies are from different host species to prevent the secondary antibody from binding to both [12].
→ Validate with Robust Controls
A well-designed experimental plan includes controls that are indispensable for diagnosing cross-reactivity.
- Negative Control: A well containing only sample matrix and secondary antibody (no primary antibody). Any signal indicates direct cross-reactivity with the sample components [17].
- Isotype Control: Using a primary antibody known to be irrelevant to the target antigen helps identify non-specific binding of the secondary antibody.
- Blocking Control: Pre-incubating the secondary antibody with the serum of the host species it was raised against should significantly reduce the signal, confirming specificity.
Experimental Protocol for Validating Secondary Antibody Specificity
This protocol provides a step-by-step methodology to empirically test a secondary antibody for cross-reactivity in your specific experimental system.
1. Coating: Dilute the target antigen to 2–10 µg/mL in an alkaline coating buffer (e.g., carbonate-bicarbonate buffer, pH 9.4). Add 100 µL per well to a 96-well microplate. Seal the plate and incubate for several hours at 37°C or overnight at 4°C [12]. 2. Blocking: Discard the coating solution and wash the plate 2-3 times with a wash buffer (e.g., PBS with 0.05% Tween-20). Add 200-300 µL of a blocking buffer (e.g., 1-5% BSA in PBS) to each well. Incubate at room temperature for 1-2 hours to cover any unsaturated binding sites [9] [12]. 3. Primary Antibody Incubation: Wash the plate as before. Prepare serial dilutions of your specific primary antibody in blocking buffer. Add 100 µL per well. Also, include your negative control wells (blocking buffer only, no primary antibody). Incubate for 1-2 hours at room temperature [9]. 4. Secondary Antibody Incubation: Wash the plate. Dilute the enzyme-conjugated secondary antibody (e.g., HRP- or AP-labeled) in blocking buffer. Add 100 µL per well to all wells, including the negative controls. Incubate for 1-2 hours at room temperature, protected from light [9] [36]. 5. Signal Detection and Analysis: Wash the plate thoroughly. Add 100 µL of a suitable substrate (e.g., TMB for HRP or pNPP for AP) to each well. Incubate until color develops and then stop the reaction if necessary. Read the absorbance immediately with a plate reader [9] [12]. 6. Data Interpretation: Compare the signal in the negative control wells to that of the test wells. A high signal in the negative control indicates significant cross-reactivity of the secondary antibody with the plate or residual sample matrix, invalidating the results.
The logical process for troubleshooting cross-reactivity based on control results is summarized below.
Research Reagent Solutions for Cross-Reactivity Mitigation
Selecting the right reagents is paramount for success. The following table details essential materials and their specific functions in minimizing cross-reactivity.
Table 2: Key Reagents for Robust Indirect ELISA
| Reagent / Material | Critical Function | Considerations for Cross-Reactivity |
|---|---|---|
| Cross-Adsorbed Secondary Antibodies | Binds specifically to primary antibody while ignoring proteins from sample species. | Verify the list of species against which the antibody has been adsorbed matches your experimental model [12]. |
| Blocking Agents (BSA, Ovalbumin) | Covers unsaturated protein-binding sites on the microplate. | Inadequate blocking is a primary cause of high background; concentration and type must be optimized [9]. |
| Wash Buffer (PBS with Detergent) | Removes unbound antibodies and sample proteins during assay steps. | The non-ionic detergent (e.g., Tween-20) is critical for reducing hydrophobic interactions [9]. |
| Matched Antibody Pairs | Used in sandwich ELISA; capture and detection antibodies target different epitopes. | Must be from different host species if using indirect detection to prevent secondary antibody cross-binding [12]. |
| Validated Assay Controls | Provides a benchmark for non-specific signal and confirms assay functionality. | A high signal in the negative control (no primary antibody) is a direct indicator of cross-reactivity [17]. |
Within the comparative framework of ELISA variants, the indirect format offers a powerful balance of sensitivity and flexibility, but this comes with an inherent responsibility to manage cross-reactivity. The selection of a highly specific, cross-adsorbed secondary antibody is the most critical factor in this endeavor. By integrating rigorous reagent selection, comprehensive control strategies, and systematic validation, researchers can effectively neutralize the threat of cross-reactivity. This ensures that the data generated, whether in basic research or critical drug development pipelines, reflects true biological signal rather than experimental artifact, thereby upholding the highest standards of scientific rigor.
The sandwich ELISA (Enzyme-Linked Immunosorbent Assay) represents one of the most sensitive and specific immunoassay formats widely deployed in research, clinical diagnostics, and drug development. Unlike direct or indirect ELISA formats, the sandwich technique employs two antibodies that bind to distinct epitopes on the target analyte, effectively "sandwiching" it between a solid-phase capture antibody and an enzyme-conjugated detection antibody. This configuration enables the specific detection of antigens in complex mixtures like serum or cell culture supernatants, making it invaluable for quantifying cytokines, biomarkers, hormones, and therapeutic proteins [65]. The exceptional utility of this method, however, is contingent upon successfully overcoming two fundamental challenges: the strategic sourcing of high-quality matched antibody pairs and the vigilant mitigation of the high-dose Hook effect. These challenges, if unaddressed, can compromise assay specificity, sensitivity, and accuracy, leading to erroneous data and incorrect conclusions.
This guide provides a systematic comparison of solutions for these challenges, offering experimental data, detailed protocols, and a curated toolkit to empower researchers in developing robust and reliable sandwich immunoassays.
Sourcing and Evaluating Matched Antibody Pairs
The Critical Role of Matched Pairs
The performance of a sandwich ELISA is fundamentally dependent on the matched antibody pair. This pair consists of a capture antibody, immobilized on a microplate well, and a detection antibody, which is conjugated to a reporter enzyme. For a successful assay, these two antibodies must bind simultaneously to the same target protein without steric hindrance. This requires them to recognize non-overlapping epitopes [65]. The use of mismatched antibodies, or those that compete for the same binding site, can result in low signal, poor sensitivity, or complete assay failure. Furthermore, the clonality of the antibodies (monoclonal vs. polyclonal) influences performance. Monoclonal antibodies offer superior batch-to-batch consistency and defined specificity, whereas polyclonal antibodies can increase the likelihood of target detection by recognizing multiple epitopes, potentially enhancing sensitivity [66] [65].
Comparison of Commercial Matched Antibody Pair Providers
Numerous commercial suppliers offer matched antibody pairs, each with distinct formulations and advantages. The table below summarizes key suppliers and their product characteristics to aid in selection.
Table 1: Comparison of Commercial Matched Antibody Pair Providers
| Supplier | Product Formats | Key Features | Species Reactivity | Validation |
|---|---|---|---|---|
| Abcam [66] | Carrier-free pairs & kits (incl. standard) | Recombinant monoclonal; BSA, glycerol, and azide-free; screened in plasma/serum | Human, Mouse, Rat, and more | Benchmarked for sensitivity/specificity |
| Cell Signaling Technology (CST) [67] | Carrier-free pairs & individual antibodies | ≥285 ready-to-ship pairs; epitope-specific; conjugation-ready | Human, Mouse, Rat, Monkey | Validated via sandwich ELISA |
| Proteintech [68] | PBS-only formulation pairs | Recombinant and monoclonal; conjugation-ready; for multiplex bead arrays | Human, Mouse, Rat, and more | Validated on proprietary ELISA platforms |
Experimental Protocol for Validating Antibody Pairs
Before committing to a large-scale study, validating a matched antibody pair is a crucial step. The following protocol outlines a standard procedure for testing a new antibody pair, incorporating orientation testing and a checkerboard titration to determine optimal conditions [65].
Procedure:
- Coating: Dilute the candidate capture antibody in a coating buffer (e.g., 0.1 M carbonate-bicarbonate buffer, pH 9.6) and add to a microplate well (e.g., 100 µL/well). Incubate overnight at 4°C.
- Washing: Wash the plate 3 times with a wash buffer (e.g., PBS with 0.05% Tween-20).
- Blocking: Add a blocking solution (e.g., 1% BSA or 5% non-fat dry milk in PBS) to all wells (e.g., 200 µL/well). Incubate for 1-2 hours at room temperature. Wash again.
- Antigen Addition: Add a dilution series of a known, purified standard antigen (e.g., from 0 to 1000 pg/mL) and a negative control to the wells. Incubate for 2 hours at room temperature. Wash.
- Detection Antibody Addition: Add the candidate detection antibody (conjugated to HRP or another enzyme) at various dilutions in a checkerboard titration pattern against the antigen concentrations. Incubate for 1-2 hours. Wash.
- Signal Detection: Add an appropriate substrate (e.g., TMB for HRP). Incubate in the dark for 10-30 minutes.
- Stop and Read: Stop the reaction with a stop solution (e.g., 1M H₂SO₄) and measure the absorbance immediately with a microplate reader.
Key Validation Checks:
- Specificity: The signal should be negligible in the negative control wells.
- Sensitivity: Determine the lowest concentration of antigen that can be reliably distinguished from the background.
- Orientation: Test the antibody pair in both possible orientations (i.e., reverse the roles of capture and detection antibodies) to identify the configuration that yields the highest signal-to-noise ratio [65].
- Hook Effect Check: Run the standard curve out to a very high antigen concentration (e.g., 10,000–100,000 pg/mL) to identify if and at what concentration the Hook effect occurs (see Section 3).
Understanding and Mitigating the High-Dose Hook Effect
Mechanism of the Hook Effect
The Hook effect (or prozone effect) is a phenomenon in one-step sandwich immunoassays where excessively high concentrations of the analyte cause a falsely low signal, leading to a significant underestimation of the true analyte concentration [69] [70]. This occurs because in a one-step protocol, the capture antibody, detection antibody, and sample analyte are all incubated simultaneously. At ultra-high analyte concentrations, the antigen binding sites on both the capture and detection antibodies become saturated independently. This prevents the formation of the essential "sandwich" complex, as the captured analyte and the detection antibody are not linked. The free, unlabeled analyte in solution effectively competes with the captured analyte for the limited number of labeled detection antibodies, resulting in fewer reporter enzymes being immobilized on the plate and a consequent reduction in signal [69]. The following diagram illustrates this mechanism across different concentration ranges.
Experimental Data and Case Studies
The Hook effect is a well-documented issue for analytes with wide physiological ranges. The table below summarizes experimental data from published research and commercial kits demonstrating this effect.
Table 2: Experimental Documentation of the Hook Effect
| Analyte | Assay System | Linear Range | Hook Effect Onset | Documented Impact |
|---|---|---|---|---|
| Type II Collagen [69] | One-step Sandwich ELISA Kit | 3.1 - 200 ng/mL | >300 ng/mL (Error Range: 300-3000 ng/mL) | Falsely low reported concentrations beyond 3000 ng/mL. |
| C-Reactive Protein (CRP) [71] | Lateral Flow Immunoassay | Up to ~50 µg/mL | >50 µg/mL | Final test-to-control ratio fails to distinguish between 50-250 µg/mL, critical for monitoring severe inflammation/sepsis. |
| Prolactin (PRL) [70] | Clinical Immunoassay | N/A | >100,000 mU/L | Falsely low PRL in patients with large prolactinomas, potentially leading to unnecessary surgery instead of medical therapy. Reported in 5.6% of macroadenomas. |
| Human Chorionic Gonadotropin (hCG) [70] | Clinical Immunoassay | N/A | >1,000,000 mIU/mL | False-negative or low results in cases of molar pregnancy, leading to misdiagnosis. |
Strategies to Overcome the Hook Effect
Researchers can employ several practical strategies to identify and prevent the Hook effect:
- Sample Dilution and Re-testing: The most straightforward and widely used method. If a high analyte concentration is suspected, the sample should be serially diluted (e.g., 1:10, 1:100, 1:1000) and re-assayed. A result that increases proportionally with dilution, rather than remaining constant, is a classic indicator of the Hook effect [69] [70]. This should be standard practice when analyzing samples from conditions known to produce very high analyte levels (e.g., large tumors, molar pregnancies, severe infections).
- Adopt a Two-Step Assay Protocol: This method physically separates the capture and detection steps. The sample is first incubated with the immobilized capture antibody. After washing away unbound material, the detection antibody is added in a second incubation step. This eliminates the competition between captured and free analyte for the detection antibody, thereby preventing the Hook effect [69]. Many commercial ELISA kits (e.g., Chondrex's two-step protocol for Cat #6018) offer this alternative.
- Kinetic Analysis: An advanced technique, particularly used in lateral flow assays, involves monitoring the real-time kinetics of test line development rather than relying on a single endpoint measurement. The rate of signal development can distinguish between true high concentrations and the Hook effect range, significantly increasing the dynamic range of the assay [71].
- Assay Design Awareness: Simply being aware of the possibility of the Hook effect is critical. Any unexpected low result in a context where a high value is clinically or experimentally plausible should be viewed with suspicion and investigated further with dilutional studies.
The Scientist's Toolkit: Essential Reagent Solutions
Successful sandwich ELISA development relies on a suite of essential reagents and tools. The following table details this core toolkit.
Table 3: Essential Research Reagent Solutions for Sandwich ELISA
| Tool / Reagent | Function | Key Considerations |
|---|---|---|
| Matched Antibody Pairs [66] [67] [68] | Core reagents for specific capture and detection of the target analyte. | Select monoclonal for consistency; polyclonal for potential sensitivity. Ensure epitopes are non-overlapping. |
| Protein Standards [66] | Quantified antigen used to generate a standard curve for extrapolating sample concentrations. | Must be highly pure and characterized. Should be the same protein used to validate the antibody pair. |
| Microplate Reader [72] | Instrument to measure the absorbance, fluorescence, or luminescence signal from the assay. | Choose readers compatible with 96- or 384-well formats and with appropriate detection modes (absorbance, fluorescence, TRF, luminescence). |
| Blocking Buffer [65] | A protein solution (e.g., BSA, casein) used to coat unused plastic surfaces to prevent non-specific binding of antibodies. | Must be IgG- and protease-free. Should be from a species that does not cross-react with detection antibodies. |
| Wash Buffer [65] | A buffered solution with a mild detergent (e.g., PBS with 0.05% Tween-20) to remove unbound reagents during washing steps. | Critical for reducing background signal. |
| Enzyme Substrate [72] | A chromogenic, fluorogenic, or luminescent compound converted by the reporter enzyme (e.g., HRP, AP) to generate a detectable signal. | TMB (colorimetric) is common. Luminescent substrates offer higher sensitivity. |
The sandwich ELISA remains a powerhouse technique in biomedical research, but its reliability is not guaranteed. This guide has detailed the two pivotal challenges: sourcing optimal matched antibody pairs and circumventing the high-dose Hook effect. By critically evaluating commercial antibody sources using checkerboard titration and validation protocols, and by proactively implementing strategies like sample dilution and two-step assays, researchers can ensure their data is both accurate and reproducible. A thorough understanding of these principles and the effective use of the available toolkit are fundamental to harnessing the full potential of the sandwich ELISA in scientific discovery and diagnostic development.
The Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technique for detecting and quantifying proteins, antibodies, and other biomolecules in research and diagnostic laboratories. Since its development in 1971 as a non-radioactive alternative to radioimmunoassays, ELISA has evolved into several distinct formats, each with unique characteristics and performance trade-offs [17] [2]. The strategic selection of an ELISA format is paramount, as it directly influences the sensitivity, specificity, reliability, and efficiency of an assay. These performance parameters ultimately determine the validity of experimental data and the success of research outcomes, particularly in critical fields like drug development and clinical diagnostics.
Sensitivity in ELISA refers to the assay's ability to accurately detect low concentrations of the target molecule, often defined by the limit of detection (LOD) [73]. Specificity, conversely, gauges how accurately the assay can distinguish the target molecule from other similar substances in the sample, thus avoiding false positives [73]. These two parameters often exist in a delicate balance; achieving optimal performance requires understanding the fundamental principles and procedural differences between the available formats. This guide provides a detailed comparison of direct, indirect, sandwich, and competitive ELISA formats, focusing on their inherent sensitivity and specificity trade-offs to inform strategic assay selection.
Comparative Analysis of ELISA Formats
The four primary ELISA formats—direct, indirect, sandwich, and competitive—differ significantly in their procedural workflow, the number of antibodies required, and their resultant performance characteristics. The selection of a particular format depends on factors including the complexity of the sample, the molecular nature of the analyte, the antibodies available, and the required levels of sensitivity and specificity [74] [17].
Direct ELISA: Simplicity and Speed
The direct ELISA is the most straightforward format, involving a single incubation step with an antigen directly immobilized on the plate and detected by an enzyme-conjugated primary antibody [12] [75]. This format offers significant advantages in terms of speed and simplicity, as it requires fewer steps and reagents, reducing both the total assay time and potential sources of error [12] [17]. Furthermore, it eliminates the risk of cross-reactivity from a secondary antibody, which can help minimize background noise [12] [75].
However, these advantages come at the cost of performance. Direct ELISA is generally the least sensitive format because there is no signal amplification step [12] [17]. The immunoreactivity of the primary antibody can also be adversely affected by the conjugation process [12]. Its application is best suited for scenarios requiring rapid antigen screening, where the target is abundant and high sensitivity is not critical, and when a conjugated primary antibody is readily available [75] [17].
Indirect ELISA: Versatility and Enhanced Sensitivity
The indirect ELISA builds upon the direct format by introducing a two-step detection process. An unlabeled primary antibody first binds to the immobilized antigen, followed by an enzyme-conjugated secondary antibody that recognizes and binds to the primary antibody [12] [34]. This configuration provides greater versatility, as the same labeled secondary antibody can be used with various primary antibodies from the same host species [12] [17]. A major benefit is that it preserves the immunoreactivity of the primary antibody since it is not modified by conjugation [12].
Most importantly, the indirect format offers significantly enhanced sensitivity. Each primary antibody contains several epitopes that can be bound by multiple labeled secondary antibodies, resulting in substantial signal amplification [12]. This makes indirect ELISA ideal for applications like antibody titer determination in serological surveys [75] [34]. The main disadvantages are the increased number of steps and the potential for higher nonspecific signal due to cross-reactivity of the secondary antibody, which necessitates careful selection of reagents and more rigorous optimization [12] [75].
Sandwich ELISA: Maximum Specificity and Sensitivity
The sandwich ELISA is often considered the gold standard for detecting antigens in complex mixtures due to its superior sensitivity and specificity. This format employs two antibodies that bind to distinct, non-overlapping epitopes on the target antigen. The capture antibody is first immobilized on the plate to selectively bind the antigen from the sample. After washing, a detection antibody is added to form an antibody-antigen-antibody "sandwich" [12] [17]. This dual antibody requirement makes the assay highly specific, as two separate binding events must occur to generate a signal [12] [74]. The sandwich format is also highly sensitive, as it efficiently captures the antigen from a solution and can be combined with indirect detection for further signal amplification [17] [34].
The key disadvantage of the sandwich ELISA is its technical demand. It requires a "matched pair" of antibodies that recognize different epitopes without interfering with each other's binding, which can involve significant development and optimization [12] [74]. It is also the most reagent-intensive and time-consuming of the standard formats [17]. This format is the preferred choice for quantifying specific antigens in complex biological samples like serum, tissue lysates, or cell culture supernatants, and is widely used in biomarker discovery and clinical diagnostics [75] [17] [76].
Competitive ELISA: For Small Analytes or Limited Antibodies
The competitive ELISA, or inhibition ELISA, operates on a different principle than the other formats. It is primarily used when the antigen is small and possesses only a single epitope, or when only one specific antibody is available [12] [34]. In one common configuration, the sample antigen and a labeled reference antigen compete for binding to a limited number of antibody binding sites immobilized on the plate [12] [75]. The key differentiator is that the signal generated is inversely proportional to the concentration of the target antigen in the sample; a high antigen concentration results in a weaker signal [75] [17].
The main advantage of this format is its ability to quantify small molecules or inhibitors that are not amenable to sandwich assays [75]. It can also be more robust when only one high-quality antibody exists for a target. However, competitive ELISA is typically less sensitive than the sandwich or indirect formats and requires very careful optimization of antigen and antibody concentrations to ensure accurate competition dynamics [75]. Its applications include detecting and quantifying small molecules like hormones and drugs, and assessing antibody neutralization in vaccine research [75] [76].
Performance Comparison Table
Table 1: Comprehensive comparison of key characteristics across the four main ELISA formats.
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Sensitivity | Low [17] | High [12] [17] | Highest [12] [17] | Moderate [75] |
| Specificity | Moderate | Moderate (risk of secondary cross-reactivity) [12] | Highest (requires two antibodies) [12] [17] | High [75] |
| Complexity / Steps | Low (fewest steps) [12] | Moderate (extra incubation step) [12] | High (most steps) [17] | Moderate to High [75] |
| Time Required | Fastest [12] | Moderate [12] | Slowest [17] | Moderate |
| Signal Amplification | No [12] | Yes (multiple secondaries bind primary) [12] | Yes (can be combined with indirect) [17] | No |
| Sample Purity Requirement | High (all proteins bind plate) [17] | High [17] | Low (suitable for crude samples) [17] [34] | Moderate |
| Antibody Requirements | Labeled primary antibody [75] | Unlabeled primary + labeled secondary [75] | Matched pair of capture & detection antibodies [12] [74] | One specific antibody [34] |
| Best For | Quick antigen checks, abundant targets | Antibody detection, general antigen detection | Complex samples, low abundance targets, high specificity needs | Small molecules, single-epitope antigens, haptens [75] [34] |
Advanced Concepts and Modern Innovations
Defining and Quantifying Performance
In practical terms, sensitivity is often quantified by the Limit of Detection (LOD), which is the lowest concentration of analyte that can be reliably distinguished from zero [73]. It is influenced by factors such as the affinity of the antibodies, the efficiency of the enzyme-substrate reaction, and the level of background noise. Specificity is determined by the antibody's unique binding site (paratope) and its affinity for the target epitope compared to other similar structures. Cross-reactivity, where an antibody binds to an unintended analyte with a similar epitope, is the primary threat to specificity and must be tested during assay validation [74] [73].
The trade-off between sensitivity and specificity is a fundamental challenge. For instance, increasing the concentration of the detection antibody to enhance sensitivity can simultaneously increase non-specific binding and background signal, thereby reducing specificity [77] [73]. Similarly, using a secondary antibody for amplification in an indirect or sandwich ELISA introduces a potential source of cross-reactivity. Therefore, optimal assay development involves a careful balancing act, often requiring empirical optimization of reagent concentrations, incubation times, and washing stringency to achieve the required performance [74].
Emerging Technologies and Future Directions
Recent technological advancements are pushing the boundaries of ELISA performance. Digital ELISA and single-molecule detection methods have dramatically improved sensitivity, enabling the detection of extremely low-abundance biomarkers previously undetectable [17]. One innovative approach, the Single-Molecule Colocalization Assay (SiMCA), uses total internal reflection fluorescence (TIRF) microscopy to visualize individual capture and detection antibodies labeled with distinct fluorophores [77]. By counting only colocalized signals, SiMCA effectively eliminates background from non-specifically bound detection antibodies, achieving a three-fold lower LOD for TNF-α compared to conventional ELISA while maintaining consistent performance in complex matrices like serum and whole blood [77].
Other significant trends include the move toward multiplexing, which allows for the simultaneous measurement of multiple analytes from a single sample volume, saving time and reagents [75] [17]. Automation and microfluidics are also transforming the field, enhancing reproducibility, throughput, and the potential for point-of-care testing [17]. Furthermore, the use of nanoparticles is being explored to enhance performance by improving the efficiency of the antibody-antigen binding and signal generation, leading to greater sensitivity and more robust quantification [76].
Experimental Protocols for Key ELISA Formats
The following section outlines the core procedural steps for setting up indirect, sandwich, and competitive ELISAs, which are the most commonly used formats in research and development.
Protocol 1: Indirect ELISA
The indirect ELISA is typically used to determine the concentration and titer of a specific antibody in a sample, such as serum from an immunized host [34].
Workflow Overview:
- Coating: Dilute the purified antigen of interest in a coating buffer (e.g., carbonate-bicarbonate buffer, pH 9.4). Add 50-100 µL per well to a 96-well microplate and incubate overnight at 4°C [12] [34].
- Blocking: Discard the coating solution and wash the plate 2-3 times with a wash buffer (e.g., PBS with 0.05% Tween-20). Add 200-300 µL of a blocking buffer (e.g., 5% non-fat dry milk or BSA in PBS) to each well to cover any remaining protein-binding sites. Incubate for 1-2 hours at room temperature [34].
- Primary Antibody Incubation: Wash the plate. Prepare serial dilutions of the sample containing the primary antibody (e.g., serum) in a diluent buffer. Add 100 µL of each dilution to the antigen-coated wells. Include a negative control (diluent only). Incubate for 1-2 hours at room temperature [34].
- Secondary Antibody Incubation: Wash the plate thoroughly to remove unbound primary antibody. Add 100 µL of an enzyme-conjugated secondary antibody (e.g., HRP-conjugated anti-species IgG) specific for the host of the primary antibody. Incubate for 1 hour at room temperature, protected from light [34].
- Detection: Wash the plate. Add 100 µL of a chromogenic substrate (e.g., TMB for HRP) to each well. Incubate until color develops (typically 5-30 minutes).
- Stop and Read: Stop the enzymatic reaction by adding 50-100 µL of a stop solution (e.g., 1M H2SO4 for TMB). Read the absorbance immediately using a microplate reader at the appropriate wavelength (e.g., 450 nm for TMB) [34] [2].
Protocol 2: Sandwich ELISA
The sandwich ELISA is used for sensitive and specific quantification of an antigen, often from a complex biological sample [34].
Workflow Overview:
- Coating: Dilute the capture antibody in a coating buffer. Add 50-100 µL per well to the plate and incubate overnight at 4°C [34].
- Blocking: Discard the coating solution, wash the plate, and add blocking buffer. Incubate for 1-2 hours at room temperature [74].
- Sample and Standard Incubation: Wash the plate. Add 100 µL of the test samples and a dilution series of the standard antigen to the wells. Incubate for 1-2 hours at room temperature (or as optimized) to allow the antigen to bind to the capture antibody [34].
- Detection Antibody Incubation: Wash the plate to remove unbound antigen and other sample components. Add 100 µL of the biotinylated or enzyme-conjugated detection antibody. Incubate for 1-2 hours at room temperature [74] [34]. Note: If the detection antibody is unlabeled, proceed to a secondary incubation step with an enzyme-conjugated antibody.
- Streptavidin-Enzyme Incubation (if using biotin): Wash the plate. Add 100 µL of streptavidin conjugated to HRP (or another enzyme). Incubate for 30-60 minutes at room temperature [74].
- Detection, Stop, and Read: Wash the plate thoroughly. Add substrate, stop the reaction, and read the absorbance as described in the indirect ELISA protocol [74].
Protocol 3: Competitive ELISA
Competitive ELISA is ideal for measuring small antigens or when only one antibody is available [34].
Workflow Overview:
- Coating: Coat the plate with a known amount of purified antigen (or the specific antibody, depending on the format) overnight at 4°C [75].
- Blocking: Block the plate as described in previous protocols.
- Competition Incubation: Pre-mix a constant, known amount of the enzyme-conjugated antibody with a series of dilutions of the sample antigen (containing the unknown) or standard antigen. Incubate this mixture for 1-2 hours to allow the competition to occur in solution [34].
- Transfer and Binding: Transfer the competition mixtures to the antigen-coated plate. The free, unbound conjugated antibody from the mixture will bind to the antigen immobilized on the plate. Incubate for 1 hour.
- Wash and Detect: Wash the plate to remove the entire competition mixture, including any antigen-antibody complexes formed in solution. Add substrate to measure the signal from the conjugated antibody that successfully bound to the plate. The signal is inversely proportional to the amount of antigen in the sample [75] [34].
Workflow Visualization
Diagram 1: Comparative workflows for the four main ELISA formats. Key differentiating steps are highlighted in red, and the final detection step is shown in green. The competitive ELISA uniquely produces a signal that is inversely proportional to the antigen concentration.
Essential Research Reagent Solutions
Successful ELISA development and execution depend on a suite of high-quality, well-validated reagents. The following table outlines the essential materials and their critical functions in the assay.
Table 2: Key reagents and materials required for ELISA development and execution.
| Reagent / Material | Function and Importance |
|---|---|
| Microplate | 96- or 384-well polystyrene plates with high protein-binding capacity and low well-to-well variation are standard. Clear plates are used for colorimetric detection, while black or white plates are for fluorescent or chemiluminescent signals [12] [2]. |
| Coating Antigen/Antibody | The purified protein or capture antibody immobilized to the solid phase. Purity and stability are crucial for efficient coating and optimal assay sensitivity [12] [74]. |
| Matched Antibody Pairs | For sandwich ELISA, a pair of antibodies that bind to distinct, non-overlapping epitopes on the target antigen without interference. This is the foundation of the assay's specificity [74] [17]. |
| Primary & Secondary Antibodies | High-affinity, well-characterized antibodies are essential. The choice between monoclonal (specificity) and polyclonal (sensitivity) impacts performance. Secondary antibodies are conjugated to an enzyme and provide signal amplification in indirect formats [12] [74]. |
| Blocking Buffer | A solution of irrelevant protein (e.g., BSA, casein, non-fat milk) or polymer used to cover all unsaturated surface-binding sites on the plate after coating, thereby minimizing non-specific binding and background noise [12] [74]. |
| Enzyme Conjugate | Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP) are the most common enzymes linked to antibodies or streptavidin. They catalyze the conversion of a substrate into a detectable product [12] [2]. |
| Chromogenic/ECL Substrate | The compound converted by the enzyme into a colored (chromogenic), fluorescent, or luminescent (ECL) product. The choice depends on the enzyme and the required sensitivity [12] [34]. |
| Wash Buffer | Typically PBS or Tris-based buffer with a detergent (e.g., Tween-20) to facilitate the removal of unbound reagents and reduce non-specific binding through rigorous washing between steps [74] [2]. |
| Stop Solution | An acidic or basic solution (e.g., sulfuric acid) used to halt the enzyme-substrate reaction at a defined endpoint, stabilizing the signal for measurement [34] [2]. |
The strategic selection of an ELISA format is a critical decision that directly governs the sensitivity, specificity, and overall success of an assay. There is no universally superior format; each offers a distinct profile of advantages and compromises. The direct ELISA provides speed and simplicity, the indirect offers versatility and amplification, the sandwich format delivers maximum sensitivity and specificity for proteins, and the competitive ELISA is uniquely suited for small molecules. This guide provides a structured framework for researchers and drug development professionals to make an informed choice. By understanding the inherent trade-offs and adhering to robust experimental protocols, scientists can effectively leverage the power of ELISA to generate reliable, high-quality data that advances scientific discovery and diagnostic development.
Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technology in biomedical research and clinical diagnostics, providing a powerful platform for detecting and quantifying proteins, antibodies, and other biomolecules. When selecting an appropriate immunoassay, researchers must navigate a complex landscape of technical considerations, balancing methodological advantages against practical constraints of time, cost, and experimental requirements. The choice between direct, indirect, sandwich, and competitive ELISA formats carries significant implications for assay sensitivity, specificity, throughput, and resource allocation [78] [9]. This guide provides an objective comparison of ELISA variants, focusing on their time and cost efficiency while maintaining scientific rigor for researchers, scientists, and drug development professionals.
The fundamental principle underlying all ELISA formats involves the specific binding of an antibody to its target antigen, followed by detection using an enzyme-conjugated reagent that produces a measurable signal. However, the strategic arrangement of these components varies considerably between formats, each employing distinct mechanisms to achieve optimal results for different experimental scenarios [34] [9]. Understanding these structural and procedural differences is essential for selecting the most efficient methodology that aligns with specific research objectives and resource constraints.
Comparative Analysis of ELISA Variants
The four primary ELISA formats—direct, indirect, sandwich, and competitive—each offer distinct advantages and limitations based on their structural configuration and detection mechanisms. Direct ELISA utilizes an antigen coated directly onto the plate surface detected by a single enzyme-conjugated primary antibody, creating the most straightforward protocol with minimal incubation steps [78] [9]. Indirect ELISA employs an unlabeled primary antibody followed by an enzyme-conjugated secondary antibody that recognizes the primary antibody, effectively amplifying the detection signal but requiring additional procedural steps [34] [9]. Sandwich ELISA captures the target antigen between two specific antibodies—a surface-bound capture antibody and a detection antibody—providing enhanced specificity and sensitivity ideal for complex samples [79] [78]. Competitive ELISA operates on an inhibition principle where samples containing the target antigen compete with a reference antigen for binding to a limited amount of antibody, making it particularly suitable for detecting small antigens with single epitopes [78] [34].
Table 1: Methodological Characteristics of Major ELISA Variants
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Antibody Requirements | Labeled primary antibody | Unlabeled primary + labeled secondary antibody | Matched antibody pair (capture + detection) | Single antibody (labeled or unlabeled) |
| Protocol Steps | 1. Antigen coating2. Blocking3. Labeled primary antibody4. Detection | 1. Antigen coating2. Blocking3. Primary antibody4. Labeled secondary antibody5. Detection | 1. Capture antibody coating2. Blocking3. Sample antigen4. Detection antibody5. Labeled secondary antibody (if needed)6. Detection | 1. Antigen or antibody coating2. Blocking3. Incubate sample with labeled antigen/antibody4. Detection |
| Time to Completion | 2-3 hours [80] | 3-6 hours [80] | 4-6 hours [9] | 3-5 hours [9] |
| Sample Compatibility | Purified antigens | Purified antigens, antibody detection | Complex samples (serum, tissue culture supernatants, tissue lysates) [34] | Small antigens, haptens, complex samples |
| Detection Mechanism | Direct antigen detection via labeled primary antibody | Indirect detection with signal amplification through secondary antibody | Antigen capture between two specific antibodies | Competition between sample antigen and reference for antibody binding |
Time and Cost Efficiency Metrics
The economic and temporal investments required for different ELISA formats vary significantly based on their reagent requirements, procedural complexity, and necessary optimization. Direct ELISA typically demands the least time investment (2-3 hours) due to fewer procedural steps but requires specifically labeled primary antibodies for each target, increasing long-term costs for multiple targets [78] [80]. Indirect ELISA offers greater reagent flexibility and cost-efficiency through the use of a single labeled secondary antibody that can be paired with various primary antibodies, though this comes with increased hands-on time and slightly longer protocols (3-6 hours) [9]. Sandwich ELISA provides exceptional sensitivity and specificity but requires carefully optimized matched antibody pairs and involves the most complex protocol, making it the most time-intensive (4-6 hours) and costly to develop [79] [9]. Competitive ELISA offers a streamlined approach for small molecules but may require specialized reagents and optimization for accurate quantification [78] [9].
Table 2: Time and Cost Efficiency Comparison of ELISA Variants
| Efficiency Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Hands-on Time | Low (minimal steps) | Moderate (additional incubation) | High (multiple incubations and washes) | Moderate (competition step) |
| Assay Development Cost | High (labeled primary antibodies) | Low (unlabeled primaries + reusable secondary) | Highest (matched antibody pairs) | Moderate (optimized competition conditions) |
| Per-Sample Cost | $$ | $ | $$$ | $$ |
| Throughput Capacity | High (quick turnaround) | High (amenable to automation) | Moderate (longer protocol) | High (suitable for screening) |
| Reagent Flexibility | Low (conjugated primaries required) | High (versatile secondary antibodies) | Low (dependent on antibody pairs) | Moderate (standardized competitors) |
| Equipment Requirements | Basic (plate reader, washer) | Basic (plate reader, washer) | Basic (plate reader, washer) | Basic (plate reader, washer) |
Experimental Protocols and Validation
Standardized Protocol Frameworks
Indirect ELISA Protocol (Adapted from JoVE): This protocol is particularly suitable for detecting specific antibodies in serum or hybridoma supernatants [34]. Begin by coating ELISA plate wells with 50-100 μL of purified antigen (2-10 μg/mL in carbonate-bicarbonate buffer, pH 9.4) followed by overnight incubation at 4°C or 1-2 hours at 37°C. Remove coating solution and block remaining protein-binding sites with 200 μL of blocking buffer (5% BSA or non-fat dry milk in PBS) for 1-2 hours at room temperature. After washing three times with PBS containing 0.05% Tween-20 (PBST), add 100 μL of serially diluted serum samples or standards and incubate for 1-2 hours at room temperature. Wash plate three times with PBST, then add 100 μL of enzyme-conjugated secondary antibody (diluted according to manufacturer's specifications) and incubate for 1-2 hours at room temperature. Following another washing step, add 100 μL of appropriate substrate (TMB for HRP, pNPP for AP) and incubate for 15-30 minutes. Stop the reaction with stop solution (e.g., 1M H₂SO₄ for TMB) and measure absorbance immediately using a microplate reader [34] [9].
Sandwich ELISA Protocol (Adapted from PMC): This protocol offers enhanced specificity for detecting antigens in complex biological samples [79] [34]. Coat wells with 100 μL of capture antibody (1-10 μg/mL in PBS) by incubating overnight at 4°C. After washing with PBST, block plates with 200-300 μL of blocking buffer (1-5% BSA in PBS) for 1-2 hours at room temperature. Wash plates three times with PBST, then add 100 μL of samples or standards diluted in appropriate buffer and incubate for 2 hours at room temperature or overnight at 4°C for low abundance targets. Following washing, add 100 μL of detection antibody (biotinylated or unlabeled) and incubate for 1-2 hours at room temperature. For indirect detection, wash plates and add 100 μL of enzyme-conjugated secondary antibody or streptavidin and incubate for 1 hour. After final washes, add 100 μL of substrate solution, incubate for 15-30 minutes, stop reaction, and read absorbance [79] [34] [9].
Method Validation and Verification Approaches
In regulated environments, distinguishing between method validation and verification becomes critical for maintaining compliance while optimizing resource allocation. Method validation represents a comprehensive process proving an analytical method is acceptable for its intended use, typically required when developing new methods or transferring methods between labs [81]. This rigorous assessment evaluates parameters including accuracy, precision, specificity, detection limit, quantitation limit, linearity, and robustness, often following established regulatory guidelines (ICH Q2(R1), USP <1225>) [81]. Method verification confirms that a previously validated method performs as expected under specific laboratory conditions, employing limited testing focused on critical parameters like accuracy, precision, and detection limits [81]. This approach is particularly valuable for implementing standardized methods while conserving resources.
For ELISA assays, validation parameters should include: dilutional linearity to assess parallelism between diluted samples and the standard curve; precision profiles determining intra-assay (repeatability) and inter-assay (reproducibility) coefficients of variation; specificity evaluation through cross-reactivity studies and spike-and-recovery experiments in the sample matrix; and sensitivity determination through limit of blank (LoB), limit of detection (LoD), and limit of quantification (LoQ) [81] [82]. For diagnostic applications, establishing the dynamic range, hook effect region, and sample stability under various storage conditions further strengthens assay robustness [82] [9].
Technical Workflow Visualization
ELISA Method Selection Algorithm
Research Reagent Solutions
Successful ELISA implementation depends on carefully selected reagents and materials that ensure assay reproducibility, sensitivity, and specificity. The following table outlines essential components for establishing robust ELISA protocols in research and diagnostic settings.
Table 3: Essential Research Reagents for ELISA Implementation
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Solid Support | 96-well or 384-well polystyrene plates [78] | Provides surface for antigen/antibody immobilization; high protein-binding capacity (>400 ng/cm²) with low well-to-well variation (<5% CV) preferred |
| Coating Reagents | Carbonate-bicarbonate buffer (pH 9.4), PBS (pH 7.4) [78] | Optimal pH conditions for passive adsorption of proteins to plastic surfaces through hydrophobic interactions |
| Blocking Agents | BSA (1-5%), non-fat dry milk (3-5%), animal sera (5-10%) [34] [9] | Covers unsaturated binding sites to minimize non-specific background; selection depends on sample type and detection system |
| Detection Enzymes | Horseradish peroxidase (HRP), Alkaline phosphatase (AP) [78] [9] | Enzyme conjugates for signal generation; HRP offers higher sensitivity while AP provides greater stability |
| Chromogenic Substrates | TMB (tetramethylbenzidine), ABTS, pNPP [34] | Converted to colored products by enzyme conjugates; TMB provides high sensitivity for HRP while pNPP is standard for AP |
| Wash Buffers | PBS with Tween-20 (0.05-0.1%) [9] | Removes unbound reagents while maintaining assay integrity; non-ionic detergents reduce non-specific binding |
| Capture/Detection Antibodies | Monoclonal/polyclinal antibodies, matched pairs [79] [34] | Critical for specificity; sandwich ELISA requires antibody pairs recognizing different epitopes |
| Reference Standards | Purified recombinant proteins, international standards [34] | Enables quantification through standard curve generation; should be identical to native protein when possible |
Discussion and Concluding Recommendations
The selection of an appropriate ELISA format represents a critical decision point that significantly impacts both the scientific validity and practical feasibility of research projects. Our analysis demonstrates that while sandwich ELISA offers superior sensitivity and specificity for complex samples, its increased time requirements and reagent costs may not be justified for all applications [79] [9]. Similarly, direct ELISA provides rapid results with minimal steps but sacrifices the signal amplification and flexibility offered by indirect detection methods [78] [9]. These trade-offs highlight the importance of aligning methodological selection with specific experimental requirements rather than defaulting to familiar protocols.
For researchers operating under significant time or budget constraints, we recommend the following evidence-based guidelines: For high-throughput antibody screening projects, indirect ELISA provides an optimal balance of cost efficiency and sensitivity, leveraging reusable secondary antibodies to minimize per-assay expenses [80] [83]. For quantification of specific antigens in complex matrices such as serum or tissue culture supernatants, sandwich ELISA delivers the requisite specificity despite higher initial development costs [79] [34]. For rapid assessment of purified antigens or when labeled primary antibodies are already available, direct ELISA offers the most time-efficient approach [78] [9]. For small molecule detection or when only one specific antibody exists, competitive ELISA represents the only viable option despite its moderate time and cost requirements [78] [34].
The strategic implementation of appropriate ELISA methodologies ultimately enhances research productivity by optimizing resource allocation while maintaining scientific rigor. By carefully considering the time and cost efficiency metrics presented in this analysis alongside specific experimental requirements, researchers can make informed decisions that maximize both scientific impact and practical feasibility across diverse applications in basic research, drug development, and clinical diagnostics.
Head-to-Head Comparison: Selecting the Right Assay for Your Research
Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technique in modern laboratories for detecting and quantifying peptides, proteins, antibodies, and hormones in biological samples [2]. Since its development in the 1970s as an alternative to radioimmunoassays, ELISA has evolved into a routine diagnostic and research method worldwide [9]. The technique utilizes the catalytic properties of enzymes to detect and quantify immunologic reactions, with components nonspecifically adsorbed or covalently bound to a solid phase like microtiter wells [9]. This solid-phase approach facilitates separation of bound and free-labeled reactants, making ELISA particularly valuable for clinical analyses [9].
The fundamental principle underlying all ELISA formats is detecting antigen-antibody interactions through enzyme-labelled conjugates and substrates that generate measurable color changes [2]. The most common enzymes used are horseradish peroxidase (HRP) and alkaline phosphatase (AP), which react with substrates like 3,3',5,5'-tetramethylbenzidine (TMB) or p-nitrophenyl-phosphate (pNPP) to produce detectable signals [34] [9]. The intensity of the color generated is proportional to the amount of analyte present in the sample, allowing for quantification against a standard curve [2].
This guide provides a comprehensive comparative analysis of the three primary ELISA formats - direct, indirect, and sandwich - to assist researchers, scientists, and drug development professionals in selecting the most appropriate methodology for their specific applications. Understanding the distinctions in detection strategies, sensitivity, procedural requirements, and optimal use cases for each format is crucial for designing robust immunoassays and accurately interpreting experimental results.
Comparative Analysis Table
The following table provides a detailed comparison of the three main ELISA types, highlighting their key characteristics, performance metrics, and optimal use cases:
Table 1: Comprehensive Comparison of Direct, Indirect, and Sandwich ELISA Methods
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA |
|---|---|---|---|
| Detection Strategy | Antigen immobilized directly on plate; detected with conjugated primary antibody [11] [9] | Antigen immobilized on plate; detected with unconjugated primary antibody followed by conjugated secondary antibody [34] [9] | Capture antibody coated on plate; antigen sandwiched between capture and detection antibodies [34] [11] |
| Number of Antibodies | One (conjugated primary antibody) [84] | Two (unconjugated primary and conjugated secondary antibody) [84] [9] | Two or more (capture antibody and conjugated detection antibody; may include secondary detection antibody) [34] [85] |
| Procedure Time | Shortest (~2-3 hours) [84] [11] | Moderate (~3-4 hours, plus additional incubation) [84] [11] | Longest (multiple incubation steps, often >4 hours) [11] [9] |
| Sensitivity | Low (minimal signal amplification) [84] [9] | High (signal amplification through multiple secondary antibodies binding to primary) [84] [11] | Highest (antigen concentrated between two antibodies) [84] [9] |
| Specificity | Lower (single antibody-epitope interaction) [11] | Moderate (potential for cross-reactivity with secondary antibody) [11] [9] | Highest (requires two distinct antibody-epitope interactions) [11] [9] |
| Complexity | Simplest protocol [84] | Moderate complexity [84] | Most complex (requires matched antibody pairs) [11] [9] |
| Sample Requirement | May require purified antigen due to direct immobilization [85] | May require purified antigen due to direct immobilization [34] | Compatible with crude/complex samples (serum, cell lysates) [34] [11] |
| Signal Amplification | None [85] | Yes (multiple secondary antibodies can bind to single primary) [11] [85] | Yes (inherent in design) [34] |
| Antibody Flexibility | Low (each primary antibody must be individually conjugated) [11] | High (same conjugated secondary can be used with various primaries) [11] [9] | Moderate (requires carefully selected antibody pairs) [11] [85] |
| Cost Considerations | Higher (due to antibody conjugation needs) [9] | Lower (versatile secondary antibodies) [9] | Highest (requires two specific antibodies per target) [9] |
| Common Applications | Assessing antibody affinity and specificity; investigating blocking/inhibitory interactions [11] | Measuring endogenous antibody levels (e.g., serological testing for HIV, autoimmune diseases) [84] [34] | Determining analyte concentration in complex biological samples; clinical diagnostics [34] [11] |
| Key Limitations | Potential for high background with complex samples; low sensitivity [11] [9] | Cross-reactivity potential from secondary antibody [11] [9] | Requires antibody pairs recognizing different epitopes; not suitable for small antigens [11] [9] |
Experimental Protocols
Direct ELISA Protocol
The direct ELISA protocol involves fewer steps than other formats, making it the most straightforward to perform [84]. The procedure begins with coating the ELISA plate with a known antibody specific to the target antigen [2]. The plate is typically incubated overnight at 4°C or for 1-2 hours at 37°C to allow passive adsorption of the antibody to the plastic surface through hydrophobic interactions [12]. After coating, the plate is washed with phosphate-buffered saline (PBS) containing a non-ionic detergent such as Tween-20 to remove unbound antibodies [9].
The next critical step is blocking, which involves adding an irrelevant protein or other molecule to cover all unsaturated surface-binding sites of the microplate wells [12]. Common blocking agents include bovine serum albumin (BSA), non-fat dry milk, or animal sera, typically incubated for at least 1-2 hours at room temperature [9]. This step is crucial to prevent nonspecific binding of detection reagents in subsequent steps, which would cause high background signal [85].
After blocking and washing, the test sample containing the antigen of interest is added to the plate and incubated, allowing the antigen to bind to the immobilized antibody [2]. The plate is washed again, and a conjugated primary detection antibody that binds directly to the target protein is added [11] [9]. This enzyme-conjugated antibody (typically using HRP or AP) is incubated for 1-2 hours, after which unbound antibody is washed away [9]. Finally, an appropriate substrate is added, and the enzymatic reaction produces a measurable color change that can be quantified using a spectrophotometer [9].
Indirect ELISA Protocol
The indirect ELISA shares initial steps with the direct format but includes an additional amplification detection step [11]. The protocol begins with coating the plate with purified antigen, typically diluted in carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4) to facilitate optimal binding to the plate surface [12] [34]. Coating is usually performed overnight at 4°C, after which the plate is washed and blocked with a protein blocking agent as described for the direct ELISA [34].
Following blocking, the test sample containing the primary antibody is added to the plate [34]. In serological applications, this typically involves serum or plasma samples from individuals suspected of having antibodies against the antigen of interest [9]. The sample is incubated for 1-2 hours at room temperature or 37°C to allow antibody-antigen binding [34]. After washing, an enzyme-conjugated secondary antibody with specificity for the primary antibody is added [11]. For example, if the primary antibody is from mouse, the secondary would be anti-mouse IgG conjugated to HRP or AP [11].
The secondary antibody incubation typically lasts 1-2 hours, after which unbound conjugate is washed away [34]. The critical advantage of this format is that multiple secondary antibodies can bind to a single primary antibody, providing signal amplification [11] [85]. Finally, substrate is added, and color development is measured [34]. The indirect ELISA protocol is particularly valuable for serological testing, such as in HIV detection, where high sensitivity is required to detect even low antibody titers [84].
Sandwich ELISA Protocol
The sandwich ELISA begins with a different approach to plate preparation compared to direct and indirect formats [9]. Rather than coating with antigen, the plate is first coated with a capture antibody specific for the target analyte [34] [11]. This initial coating step typically occurs overnight at 4°C, using antibody concentrations generally between 2-10 μg/mL in alkaline coating buffer [12]. After washing, the plate is blocked with protein blocking agent to prevent nonspecific binding [85].
The test sample containing the antigen of interest is then added to the plate [34]. Complex samples such as serum, plasma, cell culture supernatants, or tissue lysates can be used directly without purification because the capture antibody specifically immobilizes the target antigen while other components are washed away [34] [11]. The antigen incubation typically lasts 90 minutes at 37°C [9].
After washing away unbound material, a detection antibody specific to a different epitope on the antigen is added [11]. This antibody may be enzyme-conjugated directly, or it may be unconjugated followed by an enzyme-conjugated secondary antibody [34]. The use of a secondary detection antibody provides additional signal amplification but requires careful selection to avoid cross-reactivity with the capture antibody [34]. This is typically achieved by using antibodies from different host species for capture and detection (e.g., mouse IgG capture with rabbit IgG detection) [12]. After final washing, substrate is added, and color development is measured [9].
The sandwich ELISA format is particularly advantageous for analyzing complex samples because the antigen does not need to be purified before analysis, and the dual antibody requirement provides exceptional specificity [34]. This format forms the basis for many commercial ELISA kits and clinical diagnostic tests [34] [11].
Experimental Workflow Diagrams
Diagram 1: Comparative workflow of the three main ELISA types showing the stepwise procedures and required wash steps between each incubation. The direct ELISA requires the fewest steps, while sandwich ELISA involves the most complex procedure with additional antibody layers.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for ELISA Experiments
| Reagent/Material | Function/Purpose | Specifications & Considerations |
|---|---|---|
| Microplates | Solid phase for immobilizing antigens or antibodies [12] | 96-well polystyrene plates with high protein-binding capacity (>400 ng/cm²); clear for colorimetric, black/white for fluorescent detection [12] |
| Coating Antibodies | Capture target analyte in sandwich ELISA [11] | Specific to target antigen; often used at 2-10 μg/mL in carbonate/bicarbonate buffer (pH 9.4-9.6) [12] [85] |
| Detection Antibodies | Bind to captured antigen for signal generation [11] | Specific to different epitope than capture antibody; may be directly conjugated or require secondary detection [34] [11] |
| Blocking Buffers | Prevent non-specific binding [12] | BSA (1-5%), non-fat dry milk (3-5%), or animal sera in PBS or TBS; must not interfere with antibody-antigen interactions [85] [9] |
| Enzyme Conjugates | Catalyze substrate conversion to detectable signal [12] | HRP or AP conjugated to antibodies; HRP with TMB substrate (detection at 450nm) or AP with pNPP (detection at 405nm) [34] [9] |
| Wash Buffers | Remove unbound reagents between steps [9] | PBS or TBS with 0.05-0.1% Tween-20; sufficient washing critical for reducing background [34] [9] |
| Stop Solution | Terminate enzyme-substrate reaction [2] | Acidic (H₂SO₄, HCl) for HRP/TMB; alkaline (NaOH) for AP/pNPP; stabilizes signal for measurement [2] [9] |
Applications in Current Research
ELISA methodologies continue to play critical roles in diverse research applications, particularly in pharmaceutical development and clinical diagnostics. Recent studies highlight the ongoing importance of these techniques in advancing biomedical research. In therapeutic drug monitoring for inflammatory bowel disease, ELISA remains the gold standard for quantifying serum concentrations of biologic drugs like ustekinumab, with established thresholds for clinical efficacy (3.9 μg/mL for clinical remission in Crohn's disease and ≥3.7 μg/mL for clinical response in ulcerative colitis) [86] [87]. The technique's precision in quantifying drug exposure is crucial for managing interindividual variability in drug clearance and optimizing therapeutic outcomes [86].
Serological applications continue to represent a major use case for ELISA formats, particularly in infectious disease research. During the COVID-19 pandemic, in-house indirect ELISA assays demonstrated substantial agreement (80.8% concordance, κ=0.61) with commercial chemiluminescent immunoassays for detecting anti-SARS-CoV-2 antibodies, highlighting their utility as cost-effective tools for serosurveillance studies [46]. The robust performance of these assays, particularly when detecting IgG antibodies against the receptor-binding domain of the spike protein, underscores the continued value of well-optimized ELISA methodologies in public health responses to emerging pathogens [46].
The choice between direct, indirect, and sandwich ELISA formats depends heavily on the specific research requirements. Sandwich ELISA formats predominate in clinical diagnostics and commercial test kits due to their superior sensitivity and specificity, particularly when analyzing complex biological matrices [34] [11]. Indirect ELISA remains the format of choice for serological applications such as HIV and autoimmune disease testing, where signal amplification enhances detection sensitivity [84] [9]. Direct ELISA finds application in specialized contexts where antibody affinity and specificity assessments are needed, or when investigating blocking and inhibitory interactions [11].
Ongoing methodological developments continue to refine ELISA applications, including the comparison with emerging point-of-care assays that offer rapid turnaround times (15-20 minutes versus 4-8 hours for traditional ELISA) while maintaining strong correlation with established ELISA techniques [86] [87]. These advancements ensure that ELISA methodologies will remain foundational tools in biomedical research, drug development, and clinical diagnostics for the foreseeable future.
Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technique in biomedical research and clinical diagnostics, enabling the detection and quantification of soluble substances such as peptides, proteins, antibodies, and hormones [9]. Since its development in the 1970s as an alternative to radioimmunoassays, ELISA has evolved into a routine laboratory method with global applications spanning disease diagnosis, drug development, and biomedical research [2] [9]. The technique operates on the fundamental principle of detecting antigen-antibody interactions through enzyme-labelled conjugates and substrates that generate measurable color changes [2].
Among the various ELISA formats that have emerged, including direct, indirect, sandwich, and competitive protocols, significant performance variations exist, particularly regarding sensitivity and specificity [88] [9]. These differences stem from their distinct molecular architectures and detection mechanisms. This article provides a comprehensive comparative analysis of these ELISA formats, with particular emphasis on the sensitivity advantages of sandwich ELISA over direct ELISA, supported by experimental data, methodological protocols, and practical applications for research and drug development professionals.
Fundamental Principles of Major ELISA Formats
Direct ELISA: Simplicity at a Cost
The direct ELISA format represents the most straightforward approach, employing a single antibody for detection. In this protocol, the target antigen is first immobilized directly onto the microplate wells [9]. After blocking unsaturated binding sites to minimize nonspecific background, an enzyme-conjugated primary antibody specific to the antigen is added [88]. Following incubation and washing steps to remove unbound antibodies, a substrate is introduced, and the enzymatic reaction generates a measurable signal proportional to the antigen concentration [12].
The primary advantage of direct ELISA lies in its procedural simplicity and rapidity, as it requires fewer steps and reagents than other formats [42]. The direct conjugation of the enzyme to the primary antibody also eliminates potential cross-reactivity from secondary antibodies [12]. However, this format suffers from critical limitations, including limited signal amplification since only one antibody is used for detection, resulting in lower sensitivity compared to other methods [89]. Additionally, the necessity for individually conjugating each primary antibody makes the process time-consuming and expensive, while the limited availability of commercially available conjugated antibodies restricts its applicability [12] [88].
Indirect ELISA: Enhanced Signal Amplification
Indirect ELISA shares initial similarities with the direct format, beginning with antigen immobilization on the microplate [9]. The critical distinction emerges in the detection strategy, which employs two antibodies: an unlabeled primary antibody that specifically binds to the antigen, followed by an enzyme-conjugated secondary antibody that recognizes and binds to the primary antibody [88]. This layered approach provides significant signal amplification, as multiple secondary antibodies can bind to a single primary antibody, thereby enhancing detection sensitivity [29].
The indirect format offers notable practical advantages, including greater flexibility and easier antibody sourcing, as the same labeled secondary antibody can be utilized with various primary antibodies from the same host species [12]. The absence of conjugation to the primary antibody also preserves its immunoreactivity [12]. However, these benefits come with trade-offs: the additional incubation step prolongs the procedure, and the introduction of a secondary antibody increases the risk of cross-reactivity and nonspecific signal [88].
Sandwich ELISA: Superior Sensitivity and Specificity
Sandwich ELISA, the most sensitive and specific format, employs a dual-antibody system that fundamentally differs from direct and indirect approaches [9]. Rather than directly immobilizing the antigen, the process begins with coating the microplate with a capture antibody [90]. When the sample is added, the target antigen binds to this capture antibody. After washing, a second detection antibody, specific to a different epitope on the antigen, is introduced, forming the characteristic "antibody-antigen-antibody" sandwich structure [12] [88].
This format delivers superior performance through several mechanisms. The two-antibody system provides exceptional specificity by requiring recognition of two distinct epitopes, significantly reducing false positives from nonspecific binding [88]. The assay achieves high sensitivity through efficient capture and retention of the target antigen, combined with flexible detection options that can incorporate signal amplification strategies [12]. However, these advantages come with development complexities, as the method requires identifying two antibodies that recognize different, non-overlapping epitopes on the target antigen without interference [90]. The additional reagents and extended procedure also increase cost and time requirements compared to direct ELISA [88].
Comparative Sensitivity Analysis: Experimental Evidence
Side-by-Side Methodology Comparison
The sensitivity disparities between ELISA formats become evident when examining their fundamental detection methodologies. The table below summarizes the key characteristics influencing sensitivity across the three main formats:
Table 1: Sensitivity Comparison of Major ELISA Formats
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA |
|---|---|---|---|
| Signal Amplification | Limited (single antibody) | High (multiple secondary antibodies per primary) | High (dual antibody system with amplification options) |
| Background Noise | Higher [89] | Moderate | Lower |
| Dynamic Range | Limited [89] | Wider | Widest |
| Antibody Compatibility | Challenging (requires conjugated primaries) [88] | Flexible (same secondary for multiple primaries) [12] | Requires matched antibody pairs [90] |
| Detection Limit | ~ng range | ~pg-ng range | ~pg range (highest sensitivity) [12] |
| Best Applications | High-abundance antigens, rapid screening | Antibody detection, flexible applications | Low-abundance targets, complex samples [42] |
The sensitivity advantage of sandwich ELISA is further demonstrated in real-world applications. A 2025 study comparing serological assays for SARS-CoV-2 antibody detection demonstrated that an in-house sandwich ELISA showed substantial agreement (80.8% overall concordance) with the highly sensitive Elecsys CLIA method, confirming its utility for reliable detection in complex biological samples [46]. Another 2025 study comparing manual ELISA with the automated Ella system for galectin-3 detection in breast cancer patients highlighted how methodological precision directly impacts measurement reliability, with the more standardized approach showing significantly lower coefficient of variation (CV) values [91].
Molecular Mechanisms Underlying Sensitivity Differences
The superior sensitivity of sandwich ELISA stems from multiple molecular and methodological advantages. Unlike direct ELISA, which relies on a single detection event, sandwich ELISA incorporates two independent binding events (capture and detection), significantly enhancing specificity and reducing false positives from nonspecific binding [88]. The format also allows for flexible signal amplification through either direct conjugation to the detection antibody or, more commonly, through an enzyme-labeled secondary antibody that provides multiple enzyme molecules per detection antibody [12].
Additionally, the efficient antigen capture in sandwich ELISA prevents loss during washing steps, ensuring that low-concentration targets are retained for detection [12]. This is particularly crucial for complex samples like serum or plasma, where target molecules may be scarce amid numerous interfering substances [42]. The reduced hook effect in properly optimized sandwich assays also maintains linearity across a wider concentration range compared to direct ELISA, which may suffer from saturation at high antigen concentrations [12].
Experimental Protocols and Methodologies
Standardized Sandwich ELISA Protocol
The following detailed protocol for sandwich ELISA illustrates the meticulous approach required to achieve maximum sensitivity, with particular attention to critical steps that differentiate it from direct ELISA:
Plate Coating: Dilute capture antibody in carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4) to a concentration of 2-10 μg/mL. Add 100 μL per well to a 96-well polystyrene microplate [12]. Cover plate and incubate overnight at 4°C or for 1-2 hours at 37°C.
Blocking: Remove coating solution and wash plate 3 times with PBS containing 0.05% Tween-20 (PBST). Add 300 μL blocking buffer (commonly 1-5% BSA or 5% non-fat dry milk in PBS) per well. Incubate at room temperature for 1-2 hours to cover all unsaturated binding sites [9].
Antigen Incubation: Wash plate 3 times with PBST. Add 100 μL of sample or standard per well. Incubate for 90 minutes at 37°C or 2 hours at room temperature to allow antigen capture [9].
Detection Antibody Incubation: Wash plate 3-5 times with PBST. Add detection antibody (concentration determined during optimization) in blocking buffer. Incubate for 1-2 hours at room temperature [88].
Secondary Antibody Incubation (if using indirect detection): Wash plate 3-5 times with PBST. Add enzyme-conjugated secondary antibody diluted in blocking buffer. Incubate for 1-2 hours at room temperature [9].
Signal Development: Wash plate 3-5 times with PBST. Add 100 μL substrate solution (e.g., TMB for HRP, pNPP for AP). Incubate for 15-30 minutes at room temperature in the dark [9].
Reaction Termination and Reading: Stop the reaction by adding 50-100 μL stop solution (e.g., 1M H₂SO₄ for TMB). Measure absorbance immediately using a microplate reader at the appropriate wavelength (450 nm for TMB with acid stop) [2].
Critical Optimization Steps for Maximum Sensitivity
Several steps in the sandwich ELISA protocol require particular attention to achieve optimal sensitivity:
Antibody Pair Selection: The capture and detection antibodies must recognize non-overlapping epitopes on the target antigen to function effectively [90]. This often requires extensive screening or utilization of commercially validated matched pair kits.
Coating Condition Optimization: Protein binding capacity varies between plate types. Optimal coating conditions should be determined experimentally for each antibody-antigen combination, with binding capacity typically exceeding 400 ng/cm² for high-sensitivity applications [12].
Blocking Agent Selection: The choice of blocking agent (BSA, casein, non-fat dry milk, or commercial proprietary blockers) significantly impacts background signal. Testing multiple blockers during assay development is recommended to minimize nonspecific binding [9].
Incubation Time and Temperature: These parameters affect binding kinetics. While standard protocols suggest 1-2 hour incubations, extending incubation times or slightly elevating temperatures may enhance sensitivity for low-abundance targets, though this may increase overall assay time [9].
Table 2: Essential Research Reagent Solutions for ELISA Development
| Reagent Category | Specific Examples | Function | Optimization Considerations |
|---|---|---|---|
| Solid Phase | 96-well polystyrene plates [2] | Immobilizes capture antibody or antigen | Choose high binding capacity (>400 ng/cm²) with low well-to-well variation (CV <5%) [12] |
| Coating Buffers | Carbonate-bicarbonate (pH 9.4), PBS (pH 7.4) [12] | Facilitates passive adsorption to plate | Test different pH conditions for optimal protein binding |
| Blocking Agents | BSA, non-fat dry milk, casein, fish skin gelatin [9] | Covers unsaturated binding sites to reduce background | Screen multiple blockers; avoid those that may interfere with antibody-antigen interactions |
| Detection Enzymes | Horseradish Peroxidase (HRP), Alkaline Phosphatase (AP) [2] | Generates measurable signal from substrate | HRP offers higher specific activity; AP is more stable |
| Chromogenic Substrates | TMB (HRP), pNPP (AP) [9] | Enzyme substrate that produces color change | TMB offers higher sensitivity; pNPP produces linear kinetics |
| Wash Buffers | PBS with 0.05-0.1% Tween-20 [2] | Removes unbound materials between steps | Optimal detergent concentration critical for reducing background while maintaining specific binding |
Applications in Research and Drug Development
Research Applications Leveraging Sensitivity Advantages
The superior sensitivity of sandwich ELISA makes it indispensable across numerous research applications where detection of low-abundance targets is critical. In cancer biomarker research, sandwich ELISA enables quantification of low-level tumor markers like galectin-3 in breast cancer patients, with recent studies demonstrating its utility for monitoring disease progression and treatment response [91]. The format's specificity allows reliable measurement even in complex matrices like serum, where multiple interfering substances are present.
In infectious disease serology, the detection of specific antibodies at low concentrations is crucial for identifying past exposures and monitoring immune responses. A 2025 study validated an in-house indirect ELISA for detecting anti-SARS-CoV-2 RBD antibodies, demonstrating substantial agreement with commercial chemiluminescent assays, highlighting how properly optimized ELISA methods provide cost-effective alternatives without compromising detection capability [46]. The sandwich format particularly excels in cytokine and signaling molecule quantification, where targets are often present at picogram levels in complex biological fluids [2].
Drug Development and Biomarker Validation
In pharmaceutical development, sandwich ELISA plays a critical role in pharmacokinetic studies by enabling precise measurement of therapeutic protein concentrations in biological fluids throughout drug disposition studies. The format's specificity allows researchers to distinguish between administered therapeutics and endogenous counterparts, while its sensitivity permits detection across the entire therapeutic concentration range.
The technology also contributes significantly to biomarker validation, where reproducible quantification of candidate biomarkers across multiple samples is essential. The high throughput capability of sandwich ELISA, combined with its precision and sensitivity, makes it ideal for analyzing large sample sets from clinical trials [91]. Furthermore, sandwich ELISA formats have been adapted for high-throughput screening in drug discovery applications, particularly for identifying compounds that modulate protein-protein interactions or cytokine secretion [88].
Emerging Trends and Methodological Innovations
Automation and Standardization Advances
Recent years have witnessed significant advancements in ELISA technology aimed at enhancing reliability and reducing variability. Automated platforms like the Ella instrument demonstrate the trend toward standardized, minimal-handling approaches that improve precision. A 2025 comparative study revealed that automated ELISA systems showed significantly lower coefficients of variation (CV values) compared to manual methods, confirming that automation reduces human error and inter-assay variability [91]. These systems maintain the fundamental sandwich ELISA principles while incorporating microfluidic technology and automated reagent handling to achieve superior reproducibility.
Ultrasensitive Detection Methodologies
The ongoing pursuit of higher sensitivity has driven development of novel detection strategies that push beyond traditional colorimetric measurements. Digital ELISA technologies partition single enzyme molecules into microwells or droplets, enabling counting of individual molecules rather than measuring bulk signal [2]. This approach can increase sensitivity by several orders of magnitude, potentially detecting femtogram levels of target analytes.
Additionally, enhanced signal amplification systems utilizing enzyme cascades, nanoprobe technology, or chemiluminescence substrates continue to expand the detection limits of traditional sandwich ELISA [9]. These innovations maintain the fundamental sandwich architecture while augmenting signal generation and detection, particularly beneficial for applications requiring extreme sensitivity, such as early disease detection or measurement of low-abundance biomarkers in minimally invasive samples.
The sensitivity disparity between sandwich and direct ELISA formats stems from fundamental differences in their methodological approaches and molecular architectures. While direct ELISA offers simplicity and rapidity, its limited signal amplification and single-epitope recognition constrain its sensitivity and specificity. Sandwich ELISA, through its dual-antibody system, provides enhanced specificity via two independent binding events and superior sensitivity through efficient antigen capture and flexible signal amplification options.
For researchers and drug development professionals, format selection should be guided by specific application requirements. Direct ELISA may suffice for high-abundance targets requiring rapid analysis, while sandwich ELISA remains indispensable for detecting low-abundance analytes in complex matrices. Ongoing technological advancements, particularly in automation and signal detection, continue to enhance the performance and reliability of sandwich ELISA, maintaining its position as the gold standard for sensitive protein quantification in biomedical research and clinical applications.
The Enzyme-Linked Immunosorbent Assay (ELISA) represents one of the most versatile and widely utilized techniques in biomedical research and clinical diagnostics. Since its development in 1971 as a safer alternative to radioimmunoassays, ELISA has evolved into multiple distinct formats, each with unique characteristics suited to different experimental needs [9]. For researchers, scientists, and drug development professionals, selecting the appropriate ELISA format is critical for obtaining accurate, reproducible results. The choice between direct, indirect, sandwich, and competitive ELISA formats involves careful consideration of specificity, sensitivity, flexibility, and practical experimental constraints. This guide provides a comprehensive comparison of these ELISA variants, focusing on their respective strengths and weaknesses to inform assay selection and optimization.
Core Principles and Methodologies of ELISA Formats
Fundamental ELISA Mechanism
All ELISA formats share a common principle: they detect antigen-antibody interactions through enzyme-mediated signal generation. The assay relies on the immobilization of one component (antigen or antibody) to a solid surface, typically a 96-well polystyrene microplate, followed by a series of binding, washing, and detection steps [2] [12]. The key differentiator between formats lies in how the target molecule is captured and detected. The most crucial element of any ELISA is a highly specific antibody-antigen interaction, which ensures the assay's reliability and accuracy [12].
Experimental Components and Workflows
ELISA protocols share several common requirements regardless of format. Essential components include a solid phase (matrix), capture molecules (antibodies or antigens), detection conjugates (enzyme-linked antibodies), substrates that generate measurable signals, wash buffers, and stop solutions [2]. The most commonly used enzyme labels are horseradish peroxidase (HRP) and alkaline phosphatase (AP), with substrates that produce colorimetric, chemiluminescent, or fluorescent signals [12]. Between each assay step, washing with buffer is essential to remove unbound materials and reduce background signal [9].
Table 1: Essential Reagents for ELISA Development
| Reagent Category | Specific Examples | Primary Function |
|---|---|---|
| Solid Phase | 96-well polystyrene microplates | Provides surface for immobilization of capture molecules |
| Coating Reagents | Carbonate-bicarbonate buffer (pH 9.4) | Optimizes binding of antigens or antibodies to plate surface |
| Blocking Agents | BSA, ovalbumin, animal sera | Covers unsaturated binding sites to prevent nonspecific binding |
| Detection Enzymes | Horseradish peroxidase (HRP), Alkaline phosphatase (AP) | Catalyzes substrate conversion to generate measurable signal |
| Common Substrates | TMB (Tetramethylbenzidine), pNPP (p-nitrophenyl phosphate) | Produces color change upon enzyme conversion |
| Stop Solutions | Sulfuric acid, Hydrochloric acid | Halts enzyme-substrate reaction at desired timepoint |
| Wash Buffers | PBS with non-ionic detergent (e.g., Tween-20) | Removes unbound materials while maintaining protein stability |
Comparative Analysis of ELISA Formats
Direct ELISA
The direct ELISA format represents the simplest approach, where the antigen is immobilized directly onto the plate and detected using a single enzyme-conjugated primary antibody [11] [92].
Strengths and Weaknesses:
- Speed and Simplicity: With fewer steps and only one antibody required, direct ELISA offers the most straightforward protocol [11] [17].
- Elimination of Cross-Reactivity: By avoiding secondary antibodies, this format prevents potential cross-reactivity issues [12].
- Limited Sensitivity: The absence of signal amplification results in lower sensitivity compared to other formats [9] [17].
- Antibody Availability: Enzyme-conjugated primary antibodies are less commercially available, and labeling primary antibodies for each specific ELISA system is time-consuming and expensive [12].
Indirect ELISA
Indirect ELISA introduces an additional amplification step through the use of a secondary antibody. The antigen is immobilized on the plate, followed by incubation with an unlabeled primary antibody, and then an enzyme-conjugated secondary antibody that recognizes the primary antibody [27] [11].
Strengths and Weaknesses:
- Enhanced Sensitivity: Multiple secondary antibodies can bind to a single primary antibody, providing signal amplification [12] [17].
- Flexibility and Cost-Effectiveness: The same labeled secondary antibody can be used with various primary antibodies from the same host species [12] [92].
- Cross-Reactivity Risk: Secondary antibodies may cause nonspecific signal if not properly validated [9] [11].
- Extended Protocol: Additional incubation and wash steps increase total assay time [17].
Sandwich ELISA
Sandwich ELISA employs two antibodies that bind to different epitopes on the target antigen. A capture antibody is immobilized on the plate, which binds the target antigen from the sample, followed by detection with a second, enzyme-conjugated antibody [9] [11].
Strengths and Weaknesses:
- Highest Specificity and Sensitivity: The requirement for two distinct antibody-epitope interactions significantly enhances specificity, while signal amplification maintains high sensitivity [9] [12].
- Compatibility with Complex Samples: Crude samples (e.g., serum, cell lysates) can be used effectively since non-specific components are washed away [11] [17].
- Antibody Pair Optimization: Requires carefully matched antibody pairs that recognize different epitopes without interference [12] [17].
- Time and Resource Intensive: More complex development process and longer protocol duration [9] [11].
Competitive ELISA
Competitive ELISA operates on the principle of signal inhibition. The sample antigen competes with a reference antigen (often labeled) for binding to a limited amount of antibody [2] [92]. The signal generated is inversely proportional to the amount of antigen in the sample.
Strengths and Weaknesses:
- Small Molecule Detection: Ideal for quantifying small antigens with single epitopes that cannot accommodate two antibodies [12] [11].
- Reduced Sample Purification: Tolerates more crude samples with minimal processing [9].
- Inverse Signal Interpretation: Results require careful interpretation as high analyte concentration produces low signal [92] [93].
- Limited Dynamic Range: May have reduced specificity compared to sandwich formats [11].
Table 2: Comprehensive Comparison of ELISA Formats
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Sensitivity | Low | Moderate to High | Highest | Moderate to High |
| Specificity | Moderate | Moderate | Highest | Moderate |
| Assay Time | Shortest (Fewest steps) | Moderate (Extra incubation) | Longest (Multiple steps) | Moderate |
| Cost | High (Labeled primary antibodies) | Cost-effective (Reusable secondary antibodies) | High (Two specific antibodies) | Moderate |
| Sample Type | Purified antigens | Purified antigens or antibody detection | Crude, complex samples | Small molecules, low concentration antigens |
| Flexibility | Low | High | Moderate | Low |
| Signal Amplification | No | Yes | Yes | No |
| Primary Applications | Antibody affinity assessment, screening | Antibody titer measurement, research | Biomarker quantification, diagnostics | Hormone measurement, drug monitoring |
Experimental Considerations and Data Interpretation
Protocol Optimization Strategies
Successful ELISA development requires careful optimization at each stage. Coating conditions must be empirically determined for each protein/antibody pair, with typical coating concentrations ranging from 2-10 μg/mL in alkaline buffer [12]. Blocking is a critical step to prevent nonspecific binding; commonly used blocking agents include bovine serum albumin (BSA), ovalbumin, or other animal proteins [9]. For sandwich ELISA, "matched pair" antibodies must be validated to ensure they recognize different epitopes without interference [12]. Sample preparation varies by specimen type, with proper collection, protease inhibition, and minimal freeze-thaw cycles being essential for maintaining analyte integrity [27].
Data Analysis and Quality Assessment
ELISA data interpretation requires appropriate standard curve generation and statistical validation. Quantitative ELISAs utilize serial dilutions of known antigen concentrations to create standard curves, with concentration typically plotted on a log-scale x-axis and absorbance on a linear y-axis [2] [9]. The 4- or 5-parameter logistic (4PL or 5PL) curve fitting is recommended for optimal results [93]. For competitive ELISA, the standard curve is inverted, with the highest concentration corresponding to the lowest OD value [93]. Quality assessment should include calculation of the coefficient of variation (CV), with duplicates ideally within 20% of the mean [93]. Spike recovery experiments can determine matrix effects by comparing sample recovery in biological matrix versus standard diluent [93].
The choice of ELISA format depends primarily on the experimental objectives, sample characteristics, and available reagents. Sandwich ELISA provides the highest specificity and sensitivity for quantifying biomarkers in complex biological fluids, making it ideal for diagnostic applications and cytokine quantification [12] [11]. Indirect ELISA offers an excellent balance of sensitivity and flexibility, particularly for antibody detection and research applications where multiple target antigens are being studied [17]. Direct ELISA serves well for rapid screening and affinity characterization when conjugated primary antibodies are available [92]. Competitive ELISA remains the format of choice for small molecules, haptens, and cases where only a single specific antibody exists [12].
Researchers should consider that while commercial ELISA kits provide convenience, performance characteristics may vary between manufacturers. A 2017 comparative study analyzing four commercial corticosterone ELISA kits found significantly different absolute values when measuring identical serum samples, though relative differences between experimental groups remained consistent [8]. This underscores the importance of maintaining consistent methodology within a study and carefully validating any new ELISA format or kit before implementation. By understanding the fundamental strengths and limitations of each ELISA format, researchers can make informed decisions that optimize both specificity and flexibility for their particular experimental needs.
Enzyme-linked immunosorbent assays (ELISAs) are foundational tools in biomedical research and diagnostics, but selecting the optimal format is critical for assay success. This guide provides an objective comparison of direct, indirect, sandwich, and competitive ELISA variants, supported by experimental data and a practical decision matrix to inform researchers' selection process.
The core principle of ELISA involves detecting antigen-antibody interactions through enzyme-linked conjugates and substrates that generate measurable signals [2]. The four primary formats—direct, indirect, sandwich, and competitive—differ significantly in their procedural approach, sensitivity, and optimal use cases [11] [17].
Table 1: Core Characteristics of Major ELISA Formats
| Format | Principle | Key Advantage | Primary Limitation | Best Application |
|---|---|---|---|---|
| Direct ELISA | Antigen immobilized directly; single conjugated antibody detects target [11] | Fast, simple protocol with minimal steps [11] | Lower specificity and potential for high background [11] | Assessing antibody affinity; blocking/inhibitory studies [11] |
| Indirect ELISA | Antigen immobilized; primary antibody binds target, detected by conjugated secondary antibody [11] | Signal amplification increases sensitivity; flexible as same secondary can detect multiple primaries [17] [12] | Potential for cross-reactivity from secondary antibody [11] | Measuring endogenous antibody levels [11] |
| Sandwich ELISA | Capture antibody immobilized; target antigen "sandwiched" between capture and detection antibodies [94] [11] | High specificity and sensitivity; compatible with complex, unpurified samples [11] [17] | Requires matched antibody pair; more complex optimization [94] [11] | Quantifying proteins in complex biological samples [11] |
| Competitive ELISA | Sample antigen competes with labeled antigen for limited antibody binding sites [11] [12] | Effective for small molecules with single epitopes [11] [95] | Signal decreases with analyte concentration (inverse relationship) [96] | Measuring small molecules, hormones, or high-abundance antigens [11] [12] |
Performance Comparison: Experimental Data and Optimization
Sensitivity and Specificity Analysis
Sensitivity varies substantially between formats due to fundamental design differences. Sandwich ELISA typically offers the highest sensitivity due to antibody-based signal amplification, while direct ELISA provides the most basic detection approach [17]. A comparative study of β-conglycinin detection in processed foods demonstrated that sandwich ELISA formats detected much lower percentages of soy protein (0.0001%) compared to competitive formats in sausage and bread samples [97].
Specificity profiles also differ significantly. Sandwich ELISA provides superior specificity because it requires two distinct antibodies to bind different epitopes on the target antigen simultaneously [11] [17]. Competitive ELISA shows particular utility for small molecules that cannot accommodate two antibodies [11].
Table 2: Experimental Performance Metrics Across ELISA Formats
| Parameter | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Detection Limit | Moderate | High (due to amplification) | Highest | Variable (depends on assay design) |
| Dynamic Range | Narrow | Moderate | Broad | Broad for small molecules |
| Inter-Assay CV% | <15% (with optimization) | <12% (with optimization) | <10% (with optimization) | <15% (with optimization) |
| Sample Purity Requirement | High (purified antigens) | Moderate to high | Low (works with crude samples) | Moderate |
| Antibody Requirements | Labeled primary antibody | Unlabeled primary + labeled secondary | Matched antibody pair | Single antibody + labeled antigen |
Experimental Protocol Considerations
Sandwich ELISA Methodology: The generalized sandwich ELISA protocol begins with coating a microplate with capture antibody (typically 1-15 µg/mL depending on purity) [94]. After blocking with protein-based blockers (BSA or non-fat milk), samples and standards are added, allowing target antigen to bind the capture antibody [12]. A detection antibody (0.5-10 µg/mL) is then added, followed by enzyme-conjugated secondary antibody if using indirect detection [94]. Finally, substrate addition produces a measurable colorimetric, chemiluminescent, or fluorescent signal [2] [12].
Critical Optimization Steps: Checkerboard titration is essential for sandwich ELISA development, simultaneously testing various concentrations of capture and detection antibodies to identify optimal pairing [94]. For competitive ELISAs, the concentration of labeled antigen must be carefully calibrated to ensure effective competition with sample antigen [12]. Sample matrix effects must be addressed through spike-and-recovery experiments, particularly when measuring analytes in complex biological fluids [94].
Inter-Assay Variability: Comparative Data
A 2017 study comparing four commercial corticosterone ELISA kits demonstrated significant variability between different kits analyzing identical serum samples [8]. The Arbor Assays kit yielded significantly higher values (357.75 ± 210.52 ng/mL) compared to DRG-5186 (40.25 ± 39.81 ng/mL) and Enzo kits (66.27 ± 51.48 ng/mL), highlighting how format and antibody selection dramatically impact absolute concentration measurements [8]. Despite absolute value differences, correlation between kits remained high, suggesting formats can reliably determine relative differences within studies [8].
Decision Matrix: Selecting the Optimal ELISA Format
The following decision matrix provides a systematic approach for researchers to select the most appropriate ELISA format based on specific experimental requirements and sample characteristics.
Figure 1: Decision pathway for selecting appropriate ELISA format based on experimental requirements.
Essential Research Reagent Solutions
Successful ELISA implementation requires specific laboratory materials and reagents. The following table details essential components and their functions for establishing a robust ELISA workflow.
Table 3: Essential Reagents and Materials for ELISA Development
| Reagent/Material | Function | Specification Guidelines |
|---|---|---|
| Microplates | Solid phase for immobilization | 96-well polystyrene plates; protein binding capacity >400ng/cm²; low CV (<5%) [12] |
| Coating Antibodies | Capture target antigen | Concentration 1-15µg/mL; affinity-purified recommended [94] |
| Detection Antibodies | Bind captured antigen for detection | Concentration 0.5-10µg/mL; should recognize different epitope than capture antibody [94] |
| Blocking Buffers | Cover unsaturated binding sites | BSA (1-5%) or non-fat milk; prevents non-specific binding [12] |
| Enzyme Conjugates | Signal generation | HRP (20-200ng/mL) or AP (100-200ng/mL) for colorimetric detection [94] |
| Substrates | Generate measurable signal | TMB (colorimetric), ECL (chemiluminescent), or PNPP (colorimetric) depending on enzyme [2] [12] |
| Wash Buffers | Remove unbound material | PBS or Tris-based with detergent (e.g., 0.05% Tween-20) [2] |
| Stop Solution | Terminate enzyme reaction | Acidic (H₂SO₄, HCl) or basic (NaOH) solutions [2] |
ELISA format selection significantly impacts assay performance, data quality, and experimental outcomes. Sandwich ELISA provides superior sensitivity and specificity for quantifying proteins in complex samples but requires carefully optimized antibody pairs. Competitive ELISA offers the best solution for small molecules or high-abundance antigens. Direct and indirect formats serve specialized applications where speed or antibody detection are priorities. Researchers should consider their specific target analyte, sample matrix, and available reagents when applying this decision matrix to ensure optimal format selection for their experimental needs.
While direct, indirect, and sandwich ELISA formats form the foundation of enzyme-linked immunosorbent assays, advancing research demands more specialized tools. For particular challenges—such as quantifying small molecules or analyzing multiple analytes simultaneously—researchers must look to more advanced methodologies. Competitive ELISA provides a robust solution for measuring low molecular weight antigens, whereas multiplex ELISA enables high-throughput profiling of complex biological systems. This guide objectively compares the performance of these advanced formats against traditional alternatives, providing the experimental data and protocols necessary to inform assay selection for drug development and scientific research.
Decoding Competitive ELISA: Principles and Best Applications
The competitive ELISA format operates on a fundamentally different principle than immunometric assays. Instead of a direct correlation between antigen concentration and signal output, it employs a competitive binding reaction that produces an inverse relationship: higher antigen concentration in the sample yields lower detected signal [17] [98]. This format is particularly invaluable for quantifying small molecules with limited epitopes, such as hormones, peptides, and steroids, which are often difficult to capture effectively with traditional sandwich assays [12] [98].
Key Operational Principle
In a common competitive ELISA configuration, the plate is coated with a known antigen, or the sample antigen competes with a labeled antigen for binding to a limited quantity of capture antibodies [17] [2]. When the sample contains a high concentration of the target antigen, it occupies most antibody binding sites, leaving few available for the labeled antigen. After washing, the measurement of the remaining labeled antigen provides an inverse measurement of the sample antigen concentration [12] [98]. The standard curve for competitive ELISA is therefore characteristically decreasing, with the highest absorbance values corresponding to the lowest antigen concentrations [93].
Ideal Application Scenarios
- Small Molecule Quantification (<10,000 Daltons): The primary application is for antigens too small to accommodate two distinct antibodies simultaneously, such as cortisol, oxytocin, or cAMP [98].
- Measurement of High Antigen Concentrations: Effectively measures samples where antigen levels would exceed the linear range of sandwich ELISA, avoiding the hook effect [95].
- Scalability: The format is highly adaptable and can be applied as a modification of direct, indirect, or sandwich ELISA protocols depending on the specific experimental needs [17].
Understanding Multiplex ELISA: Simultaneous Multi-Analyte Profiling
Multiplex ELISA transforms traditional immunoassay capabilities by enabling the parallel measurement of multiple analytes from a single sample aliquot within one well [99] [17]. This technology typically employs microsphere- or bead-based arrays, where each set of beads is uniquely color-coded and coated with a specific capture antibody, allowing for the simultaneous detection of different targets [17].
Key Operational Principle
In a multiplex ELISA, a sample is incubated with a mixture of these distinct beads. Each bead set captures its specific target analyte, which is then detected using a cocktail of biotinylated detection antibodies followed by a streptavidin-phoroconjugate [17]. The analyzer then identifies each bead based on its color code and measures the associated fluorescence or chemiluminescence, quantifying multiple targets simultaneously [99].
Ideal Application Scenarios
- Comprehensive Biomarker Panels: Crucial for profiling complex cytokine/chemokine networks in immunology, signaling pathway phosphoproteins in drug development, or biomarker signatures for disease stratification [17].
- Limited Sample Volume Scenarios: Maximizes data generation from precious biobank samples, pediatric collections, or tumor biopsies where material is severely limited [17].
- High-Throughput Screening: Dramatically increases throughput and reduces reagent consumption and hands-on time in pharmaceutical screening and systems biology research [17].
Head-to-Head Comparison: ELISA Format Performance Metrics
Selecting the appropriate ELISA format requires careful consideration of performance characteristics relative to experimental goals. The table below summarizes key quantitative and qualitative parameters across different assay types.
Table 1: Comprehensive Comparison of ELISA Formats and Their Characteristics
| Format | Best For Antigen Size | Sensitivity | Specificity | Sample Type | Quantitative Relationship | Key Advantage |
|---|---|---|---|---|---|---|
| Direct ELISA | Medium-Large | Lower [17] | Lower [17] | Purified or enriched samples [17] | Direct | Fastest protocol; minimal steps [12] |
| Indirect ELISA | Medium-Large | Higher (signal amplification) [17] | Medium [17] | Purified or enriched samples [17] | Direct | High flexibility and sensitivity [12] |
| Sandwich ELISA | Medium-Large | Highest (two antibodies) [17] [12] | Highest (two antibodies) [17] [12] | Complex, crude samples (serum, lysates) [17] [95] | Direct | High specificity and sensitivity; ideal for complex samples [12] [95] |
| Competitive ELISA | Small (<10 kDa) [98] | High for small molecules [98] | High (one antibody) [98] | Various (serum, urine) [2] | Inverse | Only viable option for small molecules [98] |
| Multiplex ELISA | Various (panel-dependent) | Variable (can be high) [17] | High (with optimized panels) [17] | Complex, volume-limited samples [17] | Direct (per analyte) | Maximum data from minimal sample [17] |
Table 2: Throughput, Cost, and Technical Considerations
| Format | Relative Assay Time | Relative Cost per Sample | Technical Complexity | Multiplexing Capability |
|---|---|---|---|---|
| Direct ELISA | Shortest [12] | Low | Low | No |
| Indirect ELISA | Medium [17] | Low-Medium | Low | No |
| Sandwich ELISA | Long [17] | Medium | Medium | No |
| Competitive ELISA | Medium-Long [17] | Medium | Medium-High | Possible with bead arrays [17] |
| Multiplex ELISA | Varies (shorter overall) | Higher (but cost per data point is lower) | High | Yes (inherently) [17] |
Experimental Protocols and Data Interpretation
Protocol: Competitive ELISA for a Small Molecule (e.g., Cortisol)
Day 1: Plate Preparation
- Coating: Add a known concentration of cortisol-protein conjugate (e.g., cortisol-BSA) in carbonate-bicarbonate buffer (pH 9.4) to a 96-well microplate. Incubate overnight at 4°C [12].
- Blocking: Wash the plate 3 times with PBS containing 0.05% Tween 20 (PBST). Add a blocking buffer (e.g., 1% BSA in PBS) to all wells and incubate for 1-2 hours at room temperature [12].
Day 2: Assay and Detection
- Competition: Prepare a mixture of a constant, limited amount of anti-cortisol antibody with either standard cortisol solutions (for the standard curve) or test samples. Transfer the mixtures to the washed, coated plate. Incubate for 2 hours at room temperature. During this step, cortisol in the sample and the cortisol immobilized on the plate compete for the limited antibody binding sites [98].
- Detection Antibody: Wash the plate. Add an enzyme-conjugated secondary antibody specific to the host of the primary anti-cortisol antibody. Incubate for 1 hour at room temperature [17].
- Signal Development: Wash the plate. Add the enzyme substrate (e.g., TMB for HRP). Incubate for 15-30 minutes in the dark [2].
- Stop and Read: Add stop solution (e.g., 1M H₂SO₄). Measure the absorbance at 450 nm immediately [2].
Data Interpretation: Generate a standard curve by plotting the absorbance (y-axis) against the log of the known standard concentrations (x-axis). This curve will have a negative slope. The concentration of cortisol in unknown samples is determined by interpolation from this curve [93].
Protocol: Multiplex Bead-Based ELISA (e.g., Cytokine Panel)
- Bead Incubation: Mix the sample or standards with the magnetic bead sets, each coated with a capture antibody against a different cytokine (e.g., IL-6, TNF-α, IL-1β). Incubate for 1-2 hours with shaking.
- Detection: Wash the beads using a magnetic plate washer. Add a cocktail of biotinylated detection antibodies specific to the target cytokines. Incubate for 1 hour with shaking.
- Signal Amplification: Wash the beads and add streptavidin-phycoerythrin (SA-PE) conjugate. Incubate for 30 minutes with shaking.
- Reading and Analysis: Wash and resuspend the beads in reading buffer. Analyze the suspension using a multiplex array reader. The instrument identifies each bead set by its internal color code and quantifies the associated fluorescence (PE) for each analyte simultaneously.
Data Interpretation: The analyzer software generates a standard curve for each analyte in the panel and reports the concentration for each target in the sample.
Visualizing ELISA Workflows and Selection Logic
Competitive ELISA Workflow
Diagram 1: Competitive ELISA workflow showing the inverse signal relationship.
Multiplex ELISA Workflow
Diagram 2: Multiplex ELISA uses color-coded beads for simultaneous detection.
ELISA Format Selection Logic
Diagram 3: A logical workflow to guide the selection of the appropriate ELISA format.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of competitive and multiplex ELISAs requires specific, high-quality reagents. The following table details the essential components for setting up these advanced assays.
Table 3: Essential Research Reagent Solutions for Advanced ELISA
| Reagent/Material | Function in Competitive ELISA | Function in Multiplex ELISA | Critical Parameters |
|---|---|---|---|
| Coated Microplate | Solid phase for immobilizing known antigen [12] | N/A | High protein-binding capacity, low well-to-well variation (<5% CV) [12] |
| Color-Coded Beads | N/A | Solid phase for capture antibodies; enables multiplexing [17] | Distinct spectral signatures, uniform antibody coupling |
| Reference Antigen | Known quantity for competition; creates standard curve [93] | Known quantity for each analyte; creates standard curves [93] | High purity, precise concentration |
| Specific Antibody Pairs | A single, high-affinity antibody for the target [98] | Multiple matched pairs for each target analyte [17] | Minimal cross-reactivity, validated pairs |
| Biotinylated Detection Ab | Secondary detection (if indirect) [17] | Binds all targets; universal detection method [17] | Consistent biotin:antibody ratio, high activity |
| Enzyme-Streptavidin Conjugate | N/A | Binds to biotinylated Abs; universal signal generation [17] | High specific activity, minimal non-specific binding |
| Chromogenic/Fluoro. Substrate | Produces measurable color (e.g., TMB) [2] | Produces measurable signal (e.g., Phycoerythrin) [17] | High sensitivity, low background, compatible with reader |
| Plate Washer/Reader | Removes unbound material; measures absorbance [2] | Washes beads; identifies beads & measures fluorescence [17] | Consistent washing, appropriate optical filters |
The selection of an ELISA format is a critical determinant of experimental success. Competitive ELISA stands as the unequivocal choice for the precise quantification of small molecules, hormones, and drugs that lack multiple epitopes. In contrast, multiplex ELISA offers unparalleled power for comprehensive biomarker discovery and systems biology approaches, especially where sample volume is limited. While traditional direct, indirect, and sandwich assays remain foundational, understanding the specific applications, performance characteristics, and protocols of these advanced formats enables researchers and drug development professionals to design more efficient, informative, and robust experimental pipelines. The continued evolution of these technologies, including further automation and integration with digital tools, promises to expand their utility in both basic research and clinical diagnostics.
Conclusion
The choice between Direct, Indirect, and Sandwich ELISA is not one-size-fits-all but a strategic decision based on experimental goals. Direct ELISA offers speed and simplicity, Indirect ELISA provides enhanced sensitivity and flexibility, while Sandwich ELISA delivers superior specificity and is the gold standard for quantifying antigens in complex mixtures. Understanding the fundamental principles, methodological nuances, and comparative strengths of each format is crucial for robust and reproducible data. As biomedical research advances, these foundational techniques continue to evolve, with innovations in digital ELISA and multiplexing pushing the boundaries of sensitivity and throughput, ensuring their indispensable role in future diagnostics, therapeutic drug monitoring, and biomarker discovery.
References
- 1. ELISA是什么-ELISA法概述 - 赛默飞 [https://www.thermofisher.cn/cn/zh/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html/1000]
- 2. An overview of ELISA: a review and update on best laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11808753/]
- 3. 酶联免疫吸附实验(ELISA) 概述 [https://www.cellsignal.cn/applications/elisa/elisa-educational]
- 4. ELISA四大常用方法优缺点比较 [https://csmedlab.com/2019/08/06/elisa%E5%9B%9B%E5%A4%A7%E5%B8%B8%E7%94%A8%E6%96%B9%E6%B3%95%E4%BC%98%E7%BC%BA%E7%82%B9%E6%AF%94%E8%BE%83/]
- 5. 直接/间接/竞争/夹心ELISA实验原理 - 南京铭研生物 [https://www.merrybio.com.cn/blog/elisa-types.html]
- 6. 酶联免疫吸附实验(ELISA) [https://www.moleculardevices.com.cn/applications/enzyme-linked-immunosorbent-assay-elisa]
- 7. Comparison of Elisa with other commonly used Immunoassa [https://www.mybiosource.com/learn/comparison-of-elisa-with-other-commonly-used-immunoassa/]
- 8. Comparison of commercial ELISA assays for quantification ... [https://www.nature.com/articles/s41598-017-06006-4]
- 9. Enzyme Linked Immunosorbent Assay - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK555922/]
- 10. Comparison of radioimmunoassay and ELISA methods for ... [https://pubmed.ncbi.nlm.nih.gov/6350461/]
- 11. What is an ELISA & Types of ELISA Tests [https://www.rndsystems.com/resources/what-is-an-elisa-and-elisa-types]
- 12. Overview of ELISA | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 13. Comparison of in-house enzyme-linked immunosorbent ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701224001234]
- 14. Comparison of Three Commercial ELISA Kits for Detection ... [https://www.mdpi.com/1999-4915/17/5/716]
- 15. Top ELISA Technologies Companies & How to Compare ... [https://www.linkedin.com/pulse/top-elisa-technologies-companies-how-compare-them-v90ge/]
- 16. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOoq-F2Ncrw9AHt5cSsgYIfsPTplPY1mRgezNJdTlsa4TMnTKK7oI]
- 17. ELISA: The Complete Guide [https://www.antibodies.com/applications/elisa]
- 18. Sandwich ELISA protocol [https://www.abcam.com/en-us/technical-resources/protocols/sandwich-elisa]
- 19. ELISA Data Analysis | Thermo Fisher Scientific - ES [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-data-analysis.html]
- 20. Enzymes: principles and biotechnological applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4692135/]
- 21. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOorHOmu_cxn6HizNLtOwzUAs_GXrGstEWGNZ7Phz0zD2G9mT_qTn]
- 22. Establishment of a Sandwich ELISA for Detection of Pan- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12196402/]
- 23. Development of a sensitive sandwich ELISA for the ... [https://www.sciencedirect.com/science/article/pii/S0022175925002005?dgcid=rss_sd_all]
- 24. ELISA Development and Optimization [https://www.thermofisher.com/ag/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-development-optimization.html]
- 25. How To Optimize Your ELISA Experiments [https://www.ptglab.com/news/blog/how-to-optimize-your-elisa-experiments/?srsltid=AfmBOoqq9Vsv5LWZO-k61hpmxC-V41ztgs1U4TZHIESaUSF1ba--_3n7]
- 26. What is ELISA? - Principles, Applications & Advantages [https://lifesciences.danaher.com/us/en/library/elisa-overview.html]
- 27. Indirect and Direct ELISA [https://www.abcam.com/en-us/technical-resources/protocols/indirect-and-direct-elisa]
- 28. Elisa troubleshooting tips – High background - Blog [https://www.arp1.com/blog/post/elisa-troubleshooting-tips-high-background.html]
- 29. What is the Difference Between Direct and Indirect ELISA? [https://shop.surmodics.com/what-is-the-difference-between-direct-and-indirect-elisa]
- 30. How to Analyze ELISA Data and Calculate Results [https://www.bosterbio.com/blog/post/how-to-analyze-elisa-data-and-calculate-results-step-by-step-guide-with-troubleshooting-tips?srsltid=AfmBOoqEeNOcjnpFi65HLzol2D6tMPGtbQXmHPIcQP5K7U35-E1B0oAV]
- 31. Advantages of ELISA for Accurate and Accelerated Drug ... [https://www.moleculardevices.com/lab-notes/microplate-readers/advantages-of-elisa-for-accurate-and-accelerated-drug-discovery]
- 32. Basic Sandwich | Thermo Fisher Scientific - ID ELISA Protocols [https://www.thermofisher.com/id/en/home/references/protocols/cell-and-tissue-analysis/elisa-protocol/general-elisa-protocol/basic-sandwich-elisa-protocol.html]
- 33. | Abcam Sandwich ELISA protocol [https://www.abcam.com/en-us/technical-resources/guides/elisa-guide/sandwich-elisa-protocol]
- 34. Video: ELISA Assays: Indirect, Sandwich, and Competitive [https://www.jove.com/v/10496/elisa-assays-indirect-sandwich-and-competitive]
- 35. Step-by-step guide to ELISA | Mabtech [https://www.mabtech.com/knowledge-hub/step-step-guide-elisa]
- 36. Introduction to ELISA, Formats and Signal Amplification [https://www.jacksonimmuno.com/secondary-antibody-resource/immuno-techniques/elisa-guide-part-1/]
- 37. An Optimised Indirect ELISA Protocol for Detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10751243/]
- 38. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOooiRh64LnkspOlgXapzBWRfnz0XspAPmccYunUz6o0_ab0vHRgq]
- 39. Types of ELISA (Enzyme-linked Immunosorbent Assay) Tests [https://www.cellsignal.com/applications/elisa/types-of-elisa-tests?srsltid=AfmBOopaNrztlo8U4D7eVsPYgQCPUaxau2ELD-Up8XZqVu_K3tlLK9xj]
- 40. Next-Generation ELISA (ELISA 2.0) Market Research ... [https://www.insightaceanalytic.com/report/next-generation-elisa-market/3241]
- 41. Basic Sandwich ELISA Protocols [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/elisa-protocol/general-elisa-protocol/basic-sandwich-elisa-protocol.html]
- 42. ELISA Kit Selection Guide: Sandwich vs Competitive ... [https://synapse.patsnap.com/article/elisa-kit-selection-guide-sandwich-vs-competitive-vs-direct-methods]
- 43. Protocol: Sandwich ELISA (Colorimetric) – Direct Detection [https://www.novusbio.com/support/support-by-application/elisa/direct-sandwich-protocol?srsltid=AfmBOorvaId5BG6rOMbnjVL4fNh-T-4EqgMLuJZaaTAtWez0J7kJMwnb]
- 44. Comparison of Three Commercial ELISA Kits for Detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12116064/]
- 45. Development of an in-house indirect ELISA kit for the ... [https://bmcvetres.biomedcentral.com/articles/10.1186/s12917-025-04756-2]
- 46. Comparative evaluation of in-house ELISA and two ... [https://www.nature.com/articles/s41598-025-97050-y]
- 47. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOopk9CTiWEjEmr3h6rspF859lfrNuZIukDc8Alro58HuyywahfZK]
- 48. Guidelines for Preparing ELISA Standards | Boster Bio Blog [https://www.bosterbio.com/blog/post/guidelines-for-preparing-elisa-standards?srsltid=AfmBOopOj99_yoPdj_Vsqri6Gwv1zas4VlXhG0slZWbuGzhLhCQnmzCi]
- 49. Types of ELISA (Enzyme-linked Immunosorbent Assay) Tests [https://www.cellsignal.com/applications/elisa/types-of-elisa-tests?srsltid=AfmBOorpBXz7I9v-A6QBs7H1pRgemmtWkwr1tkUUG2G0sytttLkwhjwc]
- 50. Establishment of an indirect ELISA antibody detection method ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-025-00966-6]
- 51. Development and Application of Indirect ELISA for IBDV ... [https://pubmed.ncbi.nlm.nih.gov/40733489/]
- 52. Diagnostic performance of ELISA kits and expanded ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12557959/]
- 53. Sandwich ELISA development - ProteoGenix US [https://us.proteogenix.science/custom-assay-development/sandwich-elisa/]
- 54. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOopYsGTv1uy3_JZeOxbjZ6wnco4vWMFoa4oDoe2-A55igDmnfgDv]
- 55. Types of ELISA: Direct, Indirect, Sandwich, and Competitive [https://stjohnslabs.com/types-of-elisa/?srsltid=AfmBOop7YBpZXfSId68N61MOizvJ8aLXKUHaj5sgclDLcA2ACiqzIztM]
- 56. Faster ELISA with Better Results [https://www.genengnews.com/insights/faster-elisa-with-better-results/]
- 57. Development and validation of a sensitive sandwich ELISA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12013435/]
- 58. Development of a sandwich ELISA to detect circulating ... [https://www.nature.com/articles/s41598-023-44038-1]
- 59. Comparing Mass Spectrometry, ELISA, and Olink for ... [https://blog.genewiz.com/comparing-mass-spectrometry-elisa-and-olink-for-proteomics]
- 60. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOop1Mql3Mr7u90ZTKn052aNJGyJI2J16e3-hTL4tV6BXvtcGjAr3]
- 61. Overview of ELISA | Thermo Fisher Scientific - AR [https://www.thermofisher.com/tr/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 62. ELISA Development and Optimization [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-development-optimization.html]
- 63. ELISA Blocking Optimization [https://www.bosterbio.com/protocol-and-troubleshooting/picokine-elisa-optimization/blocking?srsltid=AfmBOoo2KA5s7FuRvi-w6eCt8oHw4gCIZCD0Rx6Ldf6bGim_ClgJ7OGv]
- 64. ELISA Guide; Part 3: ELISA Optimization [https://www.jacksonimmuno.com/secondary-antibody-resource/immuno-techniques/elisa-guide-part-3-elisa-optimization/]
- 65. Assay Setup: Sandwich ELISA for Allergy [https://www.jacksonimmuno.com/secondary-antibody-resource/immuno-techniques/sandwich-elisa/]
- 66. Matched antibody pairs for ELISA [https://www.abcam.com/en-us/technical-resources/product-overview/matched-antibody-pair-kits-for-elisa]
- 67. Matched Antibody Pairs - ELISA [https://www.cellsignal.com/applications/elisa/matched-antibody-pairs?srsltid=AfmBOoqYVIUKiVUm27j3GcYalhSq8Zxtae7RMx8Oxe8Oc2bESmonXObJ]
- 68. Matched Antibody Pairs [https://www.ptglab.com/products/matched-antibody-pairs/?srsltid=AfmBOooshb3ZGJ780N6Wsv3E-8CsXC3psYcJqc_v2iK0Hi9zFnIbCwmm]
- 69. Understanding the Hook Effect in a One-Step Sandwich ... [https://www.chondrex.com/scientific-articles/understanding-the-hook-effect-in-a-one-step-sandwich-elisa]
- 70. Hook Effect - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/hook-effect]
- 71. Mitigating the hook effect in lateral flow sandwich ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5839149/]
- 72. Optimizing your ELISA Assays [https://www.bmglabtech.com/en/blog/optimizing-your-elisa-assays/]
- 73. ELISA Sensitivity and Specificity [https://www.creative-diagnostics.com/ivd-materials/support/elisa-sensitivity-and-specificity.html]
- 74. ELISA Assay Development & Method Development [https://www.nebiolab.com/elisa-assay-method-development/]
- 75. Explore ELISA Types: Direct , Indirect , Sandwich , and... | Boster Bio [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa]
- 76. Elisa Assays in Pharmaceutical and Biotechnological ... [https://www.mybiosource.com/learn/elisa-assays-in-pharmaceutical-and-biotechnological-research/]
- 77. Improved immunoassay sensitivity and specificity using ... [https://www.nature.com/articles/s41467-022-32796-x]
- 78. Overview of ELISA | Thermo Fisher Scientific - DE [https://www.thermofisher.com/de/de/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 79. Comparison of double antigen sandwich and indirect enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7676215/]
- 80. ELISA, Flow, Western: Choosing the Right Assay for Your ... [https://precisionantibody.com/elisa-flow-western-choosing-the-right-assay-for-your-antibody/]
- 81. Method Validation vs Method Verification: Which is Better ... [https://www.labmanager.com/method-validation-vs-method-verification-which-is-better-for-use-in-laboratories-33982]
- 82. Precision-Driven Biomarker Validation: A Biotech ... [https://www.criver.com/eureka/precision-driven-biomarker-validation-biotech-perspective-part-i]
- 83. Why ELISAs Are Cost-Effective for Large-Scale Sample ... [https://www.arborassays.com/cost-effective-elisa-processing/?srsltid=AfmBOoq9UoxvYMD8Y3NcWQPsm5ZjX0YQLFC60bSm615olFwOa-13t-1a]
- 84. Guide to the Different Types of Elisa [https://hudsonlabautomation.com/a-guide-to-different-types-of-elisa/]
- 85. And Direct . Immunosorbent Assays Explained Indirect ELISA [https://bitesizebio.com/35291/direct-indirect-elisa-immunosorbent-assay/]
- 86. Comparative evaluation of point of care assay with ELISA ... [https://www.sciencedirect.com/science/article/abs/pii/S2444382425000665]
- 87. Comparative evaluation of point of care assay with ELISA ... [https://www.elsevier.es/es-revista-gastroenterologia-hepatologia-14-articulo-comparative-evaluation-point-care-assay-S021057052400284X?newsletter=true]
- 88. Antibodies 101: The Four ELISAs and When to Use Them [https://blog.addgene.org/antibodies-101-the-four-elisas-and-when-to-use-them]
- 89. What are the limitations of direct ELISA? [https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-limitations-of-direct-elisa]
- 90. The Benefits of Sandwich ELISA Kits [https://blog.crownbio.com/elisa-kit-benefits]
- 91. Comparative Analysis of Manual ELISA and Ella ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523246/]
- 92. Types of ELISA (Enzyme-linked Immunosorbent Assay) Tests [https://www.cellsignal.com/applications/elisa/types-of-elisa-tests?srsltid=AfmBOorXypmRuAppPy0u03DOuVSwsDuiXj_IBlHr1KY-95hpZiLq6fmU]
- 93. Data analysis for ELISA [https://www.abcam.com/en-us/technical-resources/guides/elisa-guide/calculating-elisa-data]
- 94. ELISA Development and Optimization [https://www.thermofisher.com/br/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-development-optimization.html]
- 95. A Researcher's Guide to Selecting the Right ELISA Kit [https://www.xlbiotec.com/blog/a-researchers-guide-to-selecting-the-right-elisa-kit/]
- 96. How to Analyze ELISA Data and Calculate Results [https://www.bosterbio.com/blog/post/how-to-analyze-elisa-data-and-calculate-results-step-by-step-guide-with-troubleshooting-tips?srsltid=AfmBOopuxqFnWANnT9qO1J3wwK2OWA9pSmLdD6cvCKfL2s5Bu-4QWIPT]
- 97. Development of sandwich and competitive ELISA formats ... [https://www.sciencedirect.com/science/article/abs/pii/S0956713519301343]
- 98. What are the differences between ELISA assay types? [https://www.enzo.com/note/what-are-the-differences-between-elisa-assay-types/]
- 99. Top Human ELISA Kits Companies & How to Compare ... [https://www.linkedin.com/pulse/top-human-elisa-kits-companies-how-compare-them-2025-elevatic-study-squ5f/]