ELISA Protocol Optimization: A Comprehensive Guide to Enhance Assay Performance for Researchers
This article provides a systematic guide for researchers, scientists, and drug development professionals to optimize Enzyme-Linked Immunosorbent Assay (ELISA) protocols.
ELISA Protocol Optimization: A Comprehensive Guide to Enhance Assay Performance for Researchers
Abstract
This article provides a systematic guide for researchers, scientists, and drug development professionals to optimize Enzyme-Linked Immunosorbent Assay (ELISA) protocols. Covering foundational principles, advanced methodological applications, step-by-step troubleshooting, and rigorous validation techniques, it synthesizes current best practices to address common challenges such as cross-reactivity, high background, and poor reproducibility. The content aims to empower users to achieve highly sensitive, specific, and reliable quantitation of proteins, hormones, and antibodies in diverse biological matrices, ultimately improving data quality and efficiency in both research and diagnostic settings.
Mastering ELISA Fundamentals: Principles, History, and Core Components
The Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technology in biomedical research and clinical diagnostics, having evolved from a novel immunoassay into an indispensable tool for protein quantification. First introduced in 1971 by Engvall and Perlmann as a safer alternative to radioimmunoassays, ELISA revolutionized biological detection by replacing radioactive labels with enzymes that produce measurable color changes upon substrate reaction [1] [2]. This transformation established a new paradigm for immunoassays that combined exceptional sensitivity with practical safety. The technique's enduring relevance stems from its powerful antibody-based design that delivers high specificity, sensitivity, and adaptability across diverse applications from virology to drug discovery [2] [3]. Within the context of ELISA protocol optimization research, understanding this evolutionary trajectory provides critical insights into how methodological refinements have expanded the technique's capabilities while maintaining its fundamental principles. This application note traces key historical developments, details optimized protocols, and explores emerging applications that continue to solidify ELISA's role in modern scientific workflows.
Historical Milestones and Technological Evolution
The development of ELISA represents a series of strategic innovations building upon foundational immunoassay principles. The historical trajectory reveals a consistent pattern of problem-solving that addressed limitations in safety, sensitivity, and practicality.
Key Historical Developments
Table 1: Major Historical Milestones in ELISA Development
| Year | Development | Key Innovators/Context | Significance |
|---|---|---|---|
| 1941 | Immunofluorescence | Albert H. Coons and team | Pioneered antibody labeling with fluorescent dyes for antigen visualization in tissues [1]. |
| 1960 | Radioimmunoassay (RIA) | Rosalyn Sussman Yalow and Solomon Berson | Enabled detection of minute biological substances using radioactive isotopes; posed health risks [1]. |
| 1971 | Invention of ELISA | Eva Engvall and Peter Perlmann (independently) | Introduced enzyme-based detection as safer alternative to RIA [1] [2] [3]. |
| 1976 | Competitive ELISA | Developed for human chorionic gonadotropin (hCG) detection | Allowed measurement of small molecules and hormones with high precision [1]. |
| 1977 | Sandwich ELISA | Introduced for enhanced specificity | Used capture and detection antibodies to "sandwich" antigen; ideal for complex samples [1]. |
| 1978 | Indirect ELISA | Created for human serum albumin detection | Employed secondary antibodies for signal amplification; increased sensitivity [1]. |
| 1985 | HIV Screening | First widely used HIV screening test | Critical public health milestone for controlling HIV/AIDS spread [1] [3]. |
| 1990s | Automation & Multiplexing | Introduction of robotic systems | Enabled high-throughput screening; minimized human error [1]. |
| 2000s | CLIA, ELFA, Microfluidics | Chemiluminescence and fluorescent assays | Provided higher sensitivity and portable point-of-care devices [1]. |
| 2010s | Digital ELISA, Integration | Single-molecule detection | Combined with mass spectrometry and NGS for detailed analyses [1]. |
| 2020s | Point-of-Care, COVID-19 | Pandemic-driven innovations | Rapid SARS-CoV-2 antibody detection; AI integration for data analysis [1]. |
Technological Transitions
The evolution from immunofluorescence to RIA and finally to ELISA marked a deliberate shift toward safer, more practical detection methodologies. While immunofluorescence established the principle of labeled antibodies, and RIA demonstrated exceptional sensitivity for hormone detection, the radiation hazards associated with RIA created a pressing need for alternatives [1]. The critical innovation emerged when researchers discovered that enzymes could effectively replace radioactive isotopes when chemically bound to antibodies, producing detectable signals through colorimetric, chemiluminescent, or fluorescent outputs [4].
The 1990s witnessed a transformative phase with the integration of automation and multiplexing capabilities. Automated ELISA systems featuring robot-assisted liquid handling and microplate readers significantly increased throughput while minimizing inter-assay variability [1]. Concurrently, multiplex ELISA techniques enabled simultaneous detection of multiple analytes within a single sample, dramatically enhancing data density from precious biological specimens [1]. These advancements established the technical foundation for contemporary high-throughput screening applications in drug discovery and clinical diagnostics.
Fundamental ELISA Principles and Formats
ELISA operates on the principle of detecting antigen-antibody interactions through enzyme-mediated signal amplification. The core components include a solid phase (typically 96-well microplates), capture molecules (antibodies or antigens), enzyme-labeled conjugates, and substrates that generate detectable signals [4]. The critical innovation lies in the "sorbent" nature of the assay, where antigens or antibodies adhere to plastic surfaces, and the "enzyme-linked" detection system that produces measurable outputs [4].
Comparative ELISA Formats
Table 2: Key Characteristics of Major ELISA Formats
| Format | Principle | Sensitivity | Applications | Advantages/Limitations |
|---|---|---|---|---|
| Direct ELISA | Antigen immobilized directly; single enzyme-labeled primary antibody detects target [3]. | Moderate | Antigen detection, screening [5] [6]. | Advantages: Simple, rapid, minimal cross-reactivity [6].Limitations: Lower sensitivity, primary antibody must be labeled. |
| Indirect ELISA | Antigen coated; primary antibody binds, enzyme-linked secondary antibody detects primary [4] [3]. | High | Antibody detection, serology [1] [4]. | Advantages: Signal amplification, flexible, same secondary for multiple primaries [1].Limitations: Potential cross-reactivity. |
| Sandwich ELISA | Capture antibody immobilized; antigen sandwiched between capture and detection antibodies [1] [7]. | Highest | Quantifying proteins, cytokines in complex samples [1] [5]. | Advantages: High specificity/sensitivity, no sample purification [1] [6].Limitations: Requires two epitope-specific antibodies. |
| Competitive ELISA | Sample antigen competes with labeled antigen for limited antibody binding sites [1] [3]. | High for small molecules | Hormones, small molecules, haptens [1] [5]. | Advantages: Suitable for small antigens, consistent [1] [6].Limitations: Inverse signal relationship. |
Figure 1: Workflow comparison of major ELISA formats showing key procedural differences and signal generation mechanisms.
Modern Applications and Workflow Integration
Contemporary Application Domains
ELISA maintains critical importance across diverse scientific and diagnostic domains. In infectious disease surveillance, ELISA-based tests revolutionized HIV screening beginning in 1985 and continue to serve as frontline diagnostic tools for diseases including malaria, dengue fever, and influenza [1] [3]. The COVID-19 pandemic highlighted ELISA's adaptability, with extensive application for detecting antibodies against SARS-CoV-2, supporting epidemiological studies and vaccine efficacy evaluations [1].
In drug discovery and development, ELISAs provide robust quantification of drug concentrations in biological samples, enabling crucial pharmacokinetic and pharmacodynamic studies [3]. The technique's precision supports biomarker validation in preclinical studies, particularly in fields requiring detection of subtle protein changes in complex fluids [2]. Additionally, ELISA formats remain integral to vaccine development through measurement of antibody responses, assessment of immunogenicity, and optimization of vaccine formulations [3].
The Researcher's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagent Solutions for ELISA Optimization
| Component | Function | Examples & Specifications | Optimization Considerations |
|---|---|---|---|
| Solid Phase | Matrix for analyte immobilization | 96-well microplates (polystyrene, polyvinyl) [4] | Plate binding capacity; well-to-well consistency; compatibility with detection system. |
| Capture Molecule | Binds target analyte | Coating antibodies (1-15 µg/mL depending on purity) [7] | Affinity-purified antibodies recommended for optimal signal-to-noise [7]. |
| Blocking Buffer | Prevents non-specific binding | BSA, non-fat milk, or commercial blocking solutions [5] | Concentration optimization critical; test different solutions for minimal background. |
| Detection Antibody | Binds captured analyte | Enzyme-conjugated detection antibody (0.5-10 µg/mL) [7] | Specificity for non-overlapping epitope (sandwich ELISA); concentration titration needed. |
| Enzyme Conjugate | Signal generation | HRP (20-200 ng/mL) or AP (100-200 ng/mL) conjugates [7] | Concentration depends on detection system (colorimetric, chemiluminescent, fluorescent). |
| Substrate | Enzyme substrate conversion | TMB (colorimetric), Luminol (chemiluminescent) [4] [3] | Selection based on sensitivity requirements and available detection instrumentation. |
| Wash Buffer | Remove unbound components | PBS with Tween-20 or commercial wash buffers [4] | Sufficient washes between steps; avoid over-washing that may disrupt bound complexes. |
| Stop Solution | Halts enzyme reaction | Acidic (H₂SO₄, HCl) or basic (NaOH) solutions [4] | Compatible with substrate and reading wavelength; adds reproducibility to timing. |
Detailed Experimental Protocols
Optimized Sandwich ELISA Protocol
The sandwich ELISA format provides exceptional specificity and sensitivity, making it ideal for quantifying proteins in complex biological samples [1] [6]. The following optimized protocol incorporates critical steps for robust assay performance.
Protocol Workflow
Figure 2: Detailed workflow for optimized sandwich ELISA protocol highlighting critical parameters and incubation conditions.
Stepwise Procedure
Plate Coating: Dilute capture antibody in coating buffer to optimal concentration (1-12 µg/mL for affinity-purified antibodies) [7]. Dispense 100 µL/well into 96-well microplate. Seal plate and incubate overnight at 4°C for maximum binding efficiency.
Washing: Aspirate coating solution and wash plate three times with 300 µL/well of wash buffer (typically PBS with 0.05% Tween-20). Soak wells for 1-2 minutes during each wash cycle to ensure thorough removal of unbound components [5]. After final wash, invert plate and blot against clean paper towels to remove residual liquid.
Blocking: Add 200-300 µL/well of blocking solution (e.g., 1-5% BSA or commercial blocking buffer). Incubate for 1-2 hours at room temperature with gentle shaking [8]. Blocking is critical for minimizing non-specific binding and reducing background signal.
Sample and Standard Incubation: Prepare serial dilutions of protein standard in diluent that matches sample matrix. Add 100 µL/well of standards, samples, and appropriate controls (blank, positive control). Incubate 2 hours at room temperature with gentle shaking. Include replicate wells for statistical reliability [9].
Detection Antibody Incubation: Dilute biotinylated or enzyme-conjugated detection antibody in standard diluent (optimal concentration typically 0.5-5 µg/mL for affinity-purified antibodies) [7]. After sample incubation and washing, add 100 µL detection antibody/well. Incubate 2 hours at room temperature.
Enzyme Conjugate Incubation: For biotinylated detection antibodies, prepare streptavidin-HRP conjugate dilution in standard diluent (20-200 ng/mL for colorimetric detection) [7]. Add 100 µL/well and incubate 30-60 minutes at room temperature protected from light.
Signal Development: Prepare fresh substrate solution according to manufacturer instructions. Add 100 µL substrate/well and incubate for precisely 15-30 minutes in the dark. Monitor color development for optimal signal intensity within the linear range.
Reaction Termination and Reading: Add 50-100 µL stop solution (typically 0.16M H₂SO₄ for TMB substrate) to each well. Read absorbance at 450 nm with 570 nm or 630 nm reference wavelength within 30 minutes to ensure signal stability [4] [5].
Competitive ELISA Protocol
For small molecules and haptens with single epitope sites, competitive ELISA provides superior quantification [1] [6]. The protocol modifies key steps from the sandwich approach:
Plate Coating: Coat plates with known antigen concentration (2-10 µg/mL) overnight at 4°C [6].
Competition Incubation: Mix constant amount of enzyme-labeled antibody with serial dilutions of standard or sample. Simultaneously add mixture to antigen-coated wells. Alternatively, pre-incubate sample with limited antibody before adding to antigen-coated wells.
Detection: Following competition incubation and washing, proceed directly to substrate addition (skip detection antibody step). The signal intensity is inversely proportional to analyte concentration in the sample [5].
Data Analysis: Generate standard curve with highest concentration corresponding to lowest OD value [9].
ELISA Optimization and Validation Strategies
Systematic Optimization Approaches
ELISA development requires meticulous optimization of each component to maximize assay window (difference between full signal and background) [8]. The checkerboard titration method represents the most efficient approach for simultaneous optimization of multiple parameters.
Figure 3: ELISA optimization strategy using checkerboard titration to simultaneously evaluate multiple parameters for optimal assay performance.
Critical Validation Procedures
Assay validation ensures ELISA results accurately reflect biological reality. Three essential validation approaches include:
Spike and Recovery: Assess matrix effects by adding known analyte amounts to both sample matrix and standard diluent. Compare quantified values after ELISA completion. Ideal recovery ranges between 80-120%; significant deviations indicate matrix interference requiring diluent modification [8] [9].
Dilutional Linearity: Serially dilute high-concentration sample beyond the standard curve's lower limit. Calculate observed versus expected concentrations after accounting for dilution factors. Proper linearity (%CV <15%) confirms consistent antibody affinity across analyte concentrations [8] [5].
Parallelism: Evaluate potential differences in antibody binding affinity between endogenous analyte and standard curve analyte. Serially dilute samples with naturally high analyte concentration and analyze against standard curve. Acceptable parallelism demonstrates consistent %CV across dilutions, validating standard curve applicability to biological samples [8].
Data Analysis and Quality Control
Standard Curve Generation and Analysis
Quantitative ELISA relies on accurate standard curve generation using serial dilutions of known analyte concentrations. Key considerations include:
- Dilution Scheme: Prepare 2-fold, 3-fold, or 5-fold serial dilutions covering the expected sample concentration range. Include at least 6-8 standard points plus blank [5].
- Replicate Measurements: Run standards and samples in duplicate or triplicate to assess precision and identify pipetting errors [9].
- Curve Fitting Models: The 4-parameter logistic (4PL) model typically provides optimal fit for sigmoidal ELISA standard curves:
- Quality Metrics: Standard curves should demonstrate R² > 0.98 for reliable quantification. Samples with OD values outside the standard curve range require dilution or concentration [5].
Troubleshooting Common ELISA Issues
Table 4: ELISA Troubleshooting Guide for Common Experimental Issues
| Problem | Potential Causes | Solutions | Preventive Measures |
|---|---|---|---|
| Low OD/No Signal | Expired substrate; inadequate incubation; over-washing; low antibody concentration [5]. | Use fresh substrate; validate incubation conditions; optimize wash stringency; titrate antibodies [5]. | Implement reagent QC; establish optimized protocol; use timer for incubations. |
| High Background | Incomplete washing; excessive detection antibody; inadequate blocking; non-specific binding [5]. | Increase wash cycles/volume; dilute detection antibody; optimize blocking solution; include negative controls [5]. | Test multiple blocking buffers; optimize antibody concentrations; validate wash efficiency. |
| High Variation Between Replicates | Inconsistent pipetting; plate sealing issues; temperature gradients; sample precipitation [9]. | Calibrate pipettes; ensure proper seal; use stable incubation environment; mix samples thoroughly [9]. | Train on pipetting technique; use quality seals; pre-warm reagents; mix samples before addition. |
| Poor Standard Curve Fit | Improper standard dilution; inadequate curve range; uneven coating; enzyme instability [5]. | Verify dilution calculations; expand standard range; ensure consistent coating; use fresh conjugates [5]. | Prepare fresh standard dilutions; include points at curve extremes; validate coating consistency. |
Emerging Trends and Future Perspectives
ELISA technology continues to evolve with several transformative trends shaping its future applications. Digital ELISA platforms now enable single-molecule detection, dramatically improving sensitivity for low-abundance biomarkers [1]. Integration with microfluidics has miniaturized traditional ELISA formats into portable, point-of-care devices suitable for resource-limited settings [1]. The COVID-19 pandemic accelerated development of rapid ELISA-based tests for SARS-CoV-2 antibodies and antigens, demonstrating the methodology's adaptability to emerging public health threats [1].
Automation and artificial intelligence integration represent the next frontier in ELISA evolution. Automated systems with robotic liquid handling and advanced microplate readers now facilitate high-throughput screening while minimizing inter-assay variability [1]. Machine learning algorithms enhance data analysis through improved curve-fitting and outlier detection capabilities [1]. These advancements complement rather than replace traditional ELISA approaches, instead expanding the technique's applicability to increasingly complex research questions.
Despite the emergence of alternative proteomic technologies, ELISA maintains distinct advantages in scenarios requiring quantitative accuracy, regulatory compliance, and practical accessibility [2]. The methodology's established infrastructure, well-characterized validation parameters, and compatibility with clinical diagnostic frameworks ensure its continued relevance. Future developments will likely focus on enhancing multiplexing capabilities, reducing sample volume requirements, and integrating with complementary analytical platforms to address increasingly sophisticated research and diagnostic challenges.
The Enzyme-Linked Immunosorbent Assay (ELISA) is a foundational technique in biomedical research and diagnostic development, leveraging the specificity of antigen-antibody interactions coupled with enzymatic amplification for detection. For researchers and drug development professionals, a deep understanding of these core principles is not merely academic; it is a prerequisite for rigorous assay optimization, reliable data interpretation, and robust diagnostic or therapeutic outcomes. The fundamental process involves immobilizing an antigen on a solid surface, complexing it with an antibody linked to an enzyme, and then measuring the enzyme's activity via incubation with a substrate to produce a quantifiable product [7]. The exquisite specificity of the assay is governed by the molecular dynamics of immunochemical binding, while its sensitivity is derived from the catalytic power of the signal-generating enzyme. This application note delineates the underlying mechanisms and provides detailed protocols for their practical application in optimizing ELISA performance.
Molecular Basis of Antigen-Antibody Interactions
Structural Foundations of Specificity
The precise interaction between an antibody and its target antigen is a paradigm of molecular recognition. The binding site is formed by the hypervariable regions of the antibody's heavy and light chains, more accurately termed Complementarity-Determining Regions (CDRs). Each antibody possesses three CDRs per chain (CDR1, CDR2, and CDR3), which create a unique molecular surface complementary to the shape and chemical character of its specific antigenic determinant, or epitope [10]. The most significant diversity is concentrated in the CDR3 region, which plays a dominant role in defining binding specificity. The combination of heavy and light chain CDRs determines the final antigen specificity, a mechanism known as combinatorial diversity [10].
Chemical Forces and Binding Dynamics
Antigen-antibody binding is a reversible, non-covalent process mediated by a combination of weak chemical forces operating over extremely short ranges [11] [10]. The contribution of each force varies depending on the specific antibody-antigen pair.
- Electrostatic Interactions: These occur between charged amino acid side chains, forming salt bridges that can be disrupted by high salt concentrations or extreme pH [10].
- Hydrogen Bonds: These bonds bridge oxygen and/or nitrogen atoms, accommodating specific reactive groups and strengthening the overall interaction [11] [10].
- Van der Waals Forces: These short-range forces operate between closely fitting surfaces, requiring a tight complementary "lock-and-key" fit [11].
- Hydrophobic Interactions: These are formed when two hydrophobic surfaces come together to exclude water. The binding energy is proportional to the surface area hidden from water [10].
The strength of an individual antibody-antigen interaction is termed its affinity, often represented by the equilibrium dissociation constant (Kd). A lower Kd indicates a higher affinity and a stronger interaction [11]. In ELISA, where antibodies are often multivalent, the overall strength of binding is referred to as avidity, which is the cumulative effect of multiple affinity interactions.
Table 1: Chemical Forces in Antigen-Antibody Interactions
| Force Type | Molecular Basis | Impact on Binding | Susceptible Disruption Method |
|---|---|---|---|
| Electrostatic | Attraction between oppositely charged ionic groups | Strong, long-range interaction; contributes significantly to specificity | High salt concentration, extreme pH |
| Hydrogen Bonding | Sharing of hydrogen between electronegative atoms | Strengthens binding; requires precise alignment of groups | Extreme pH, denaturing agents |
| Van der Waals | Fluctuating electrical charges between adjacent atoms | Very short-range; requires close surface complementarity | Detergents |
| Hydrophobic | Interaction of non-polar groups in aqueous solution | Strong contributor to binding energy; proportional to buried surface area | Detergents, organic solvents |
Principles of Enzymatic Detection
The Enzyme Conjugate as a Signal Amplifier
The detection moiety of an ELISA is typically an enzyme conjugated to a detection antibody. Common enzymes include Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP). These enzymes act as powerful amplifiers; a single enzyme molecule can catalyze the conversion of many substrate molecules to a detectable product, thereby greatly enhancing the assay's sensitivity [7]. The choice of enzyme depends on the required sensitivity, the detection method, and the need to avoid endogenous enzyme activity in the sample.
Substrate Systems and Detection Modalities
The substrate chosen for the enzyme must yield a measurable signal. The most common detection modes are absorbance (colorimetric), fluorescence, and luminescence [3]. Colorimetric substrates are widely used due to their simplicity and cost-effectiveness, with absorbance read on a standard microplate reader. Fluorescence and chemiluminescence detection generally offer higher sensitivity [3].
Table 2: Common Enzyme-Substrate Systems in ELISA
| Enzyme | Common Substrates | Detection System | Typical Conjugate Concentration |
|---|---|---|---|
| Horseradish Peroxidase (HRP) | TMB (Tetramethylbenzidine), ABTS | Colorimetric | 20–200 ng/mL [7] |
| Horseradish Peroxidase (HRP) | Enhanced Luminol | Chemiluminescent | 10–100 ng/mL [7] |
| Alkaline Phosphatase (AP) | pNPP (p-Nitrophenyl Phosphate) | Colorimetric | 100–200 ng/mL [7] |
| Alkaline Phosphatase (AP) | CDP-Star, CSPD | Chemiluminescent | 40–200 ng/mL [7] |
Experimental Protocol: Checkerboard Titration for ELISA Optimization
A critical step in developing a robust in-house ELISA, particularly in the sandwich format, is the optimization of key reagent concentrations. The checkerboard titration is an efficient experimental design that allows for the simultaneous optimization of two parameters, such as capture and detection antibody concentrations [7] [8].
1. Primary Objective: To determine the optimal concentrations of the capture antibody and detection antibody that yield a strong specific signal with low background noise.
2. Materials and Reagents:
- Coating Buffer: (e.g., 0.2 M carbonate-bicarbonate buffer, pH 9.4)
- Wash Buffer: (e.g., PBS or Tris-buffered saline with 0.05% Tween 20)
- Blocking Buffer: (e.g., 1-5% BSA or non-fat dry milk in wash buffer)
- Capture Antibody: Serial dilutions prepared in coating buffer.
- Detection Antibody: Serial dilutions prepared in standard diluent/blocking buffer.
- Antigen: A known positive control sample at a medium concentration.
- Enzyme Conjugate: (e.g., Streptavidin-HRP, if using a biotinylated detection antibody).
- Substrate: Appropriate to the enzyme (e.g., TMB for HRP).
- Stop Solution: (e.g., 0.16 M sulfuric acid for TMB).
- 96-well microplate and microplate reader.
3. Procedural Workflow:
4. Detailed Methodology:
Step 1: Plate Coating (Variable: Capture Antibody Concentration)
- Prepare a dilution series of the capture antibody in coating buffer. The range should be based on the antibody type (see Table 3). For example, prepare concentrations of 1, 2, 4, 8, and 12 µg/mL for an affinity-purified monoclonal antibody [7].
- Dispense each concentration of capture antibody in a column of the 96-well plate (e.g., Column 1: 1 µg/mL, Column 2: 2 µg/mL, etc.). Include a control well with coating buffer only.
- Seal the plate and incubate overnight at 4°C or for 1-2 hours at 37°C.
Step 2: Washing
- Aspirate the coating solution and wash the plate 3-5 times with wash buffer (200-300 µL per well per wash) to remove unbound antibody.
Step 3: Blocking
- Add a sufficient volume (e.g., 200 µL) of blocking buffer to all wells.
- Incubate for 1-2 hours at room temperature or 37°C.
- Wash the plate as in Step 2.
Step 4: Antigen Incubation
- Add a fixed, known concentration of the target antigen (your positive control) to all wells. A volume of 100 µL is standard.
- Incubate for a fixed time (e.g., 2 hours at room temperature).
- Wash the plate as before.
Step 5: Detection Antibody Incubation (Variable: Detection Antibody Concentration)
- Prepare a dilution series of the detection antibody in blocking buffer. For an affinity-purified antibody, a range of 0.5 - 5 µg/mL is a good starting point [7].
- Dispense each concentration of detection antibody in a row of the plate (e.g., Row A: 0.5 µg/mL, Row B: 1 µg/mL, etc.).
- Incubate for 1-2 hours at room temperature.
- Wash the plate thoroughly.
Step 6: Enzyme Conjugate Incubation
- If the detection antibody is not directly conjugated, add the appropriate enzyme-conjugated secondary antibody or Streptavidin (if the detection antibody is biotinylated) at the manufacturer's recommended dilution or an optimized concentration (see Table 2).
- Incubate for 30-60 minutes at room temperature.
- Wash the plate exhaustively.
Step 7: Signal Development
- Add the enzyme substrate (e.g., 100 µL of TMB) to all wells.
- Incubate in the dark for a fixed period (e.g., 15-30 minutes).
- Stop the reaction by adding an equal volume of stop solution (e.g., 100 µL of 0.16 M H₂SO₄ for TMB).
Step 8: Data Analysis
- Read the absorbance immediately on a microplate reader at the appropriate wavelength (e.g., 450 nm for TMB).
- The optimal concentration pair is the one that gives the highest signal for the positive antigen sample with the lowest signal in the negative control (no antigen) wells, resulting in the highest signal-to-noise ratio.
Table 3: Recommended Antibody Concentration Ranges for Optimization
| Antibody Source | Coating Antibody Range (µg/mL) | Detection Antibody Range (µg/mL) |
|---|---|---|
| Polyclonal Serum | 5–15 | 1–10 [7] |
| Crude Ascites | 5–15 | 1–10 [7] |
| Affinity-Purified Polyclonal | 1–12 | 0.5–5 [7] |
| Affinity-Purified Monoclonal | 1–12 | 0.5–5 [7] |
Assay Validation and Data Analysis
Critical Validation Experiments
Once optimal conditions are identified, the assay must be validated to ensure accuracy and reliability for its intended use.
- Spike and Recovery: Assess the impact of the sample matrix (e.g., serum, cell lysate) on the detection of the analyte. A known amount of analyte is spiked into the sample matrix and a reference diluent. The recovery is calculated by comparing the measured concentration to the expected concentration. Ideal recovery is 80-120% [8] [9].
- Dilutional Linearity: Evaluate the assay's performance across the expected concentration range. A sample with a high analyte concentration is serially diluted. The measured concentrations should be proportional to the dilution factor, demonstrating that the assay accurately quantifies the analyte at different levels [8].
- Parallelism: This confirms that the antibody binding affinity is the same for the endogenous analyte in the sample and the purified standard used for the calibration curve. Serially dilute a sample with a high endogenous level of the analyte. The calculated concentrations after dilution should have a low coefficient of variation (%CV) [8].
Data Analysis and Standard Curve Fitting
For quantitative ELISAs, data are interpreted using a standard curve. The mean absorbance of duplicate or triplicate standards is plotted against their known concentrations [9]. The most appropriate curve-fitting model should be selected for optimal accuracy.
- Linear and Semi-log Plots: Simple but may compress data at the curve's extremes.
- 4- or 5-Parameter Logistic (4PL/5PL): These are the gold standard for immunoassays. The 4PL model assumes symmetry around the inflection point, while the 5PL accounts for asymmetry, often providing a superior fit [9].
The concentration of unknown samples is determined by interpolating their mean absorbance from the standard curve. Any sample falling outside the range of the standard curve should be re-assayed at an appropriate dilution [9].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents for ELISA Development and Optimization
| Reagent / Material | Function / Role | Key Considerations |
|---|---|---|
| Matched Antibody Pairs | A pair of monoclonal or polyclonal antibodies that bind distinct, non-overlapping epitopes on the target antigen for sandwich ELISA [7]. | Essential for specificity; requires empirical testing for pair compatibility. |
| Blocking Buffers (e.g., BSA, Casein) | Proteins or mixtures used to coat all remaining protein-binding sites on the plate after coating with the capture antibody, minimizing non-specific binding [12]. | Different blockers may be optimal for different antibody-antigen systems; testing is required to minimize background. |
| Microplates | Solid phase for immobilization of the capture antibody or antigen. | High protein-binding plates (e.g., polystyrene) are standard. |
| Enzyme Conjugates (HRP, AP) | Catalyze the conversion of a substrate into a detectable signal, providing assay sensitivity through amplification. | Concentration must be optimized to balance signal and background (see Table 2). |
| Chromogenic/Luminescent Substrates | Molecules converted by the enzyme conjugate to a colored, fluorescent, or luminescent product for detection. | Choice depends on required sensitivity and available detection instrumentation [3]. |
| Sample/Diluent Matrix | The solution used to dilute samples and standards. | Should mimic the sample matrix as closely as possible to avoid matrix effects (e.g., using serum diluent for serum samples) [7] [9]. |
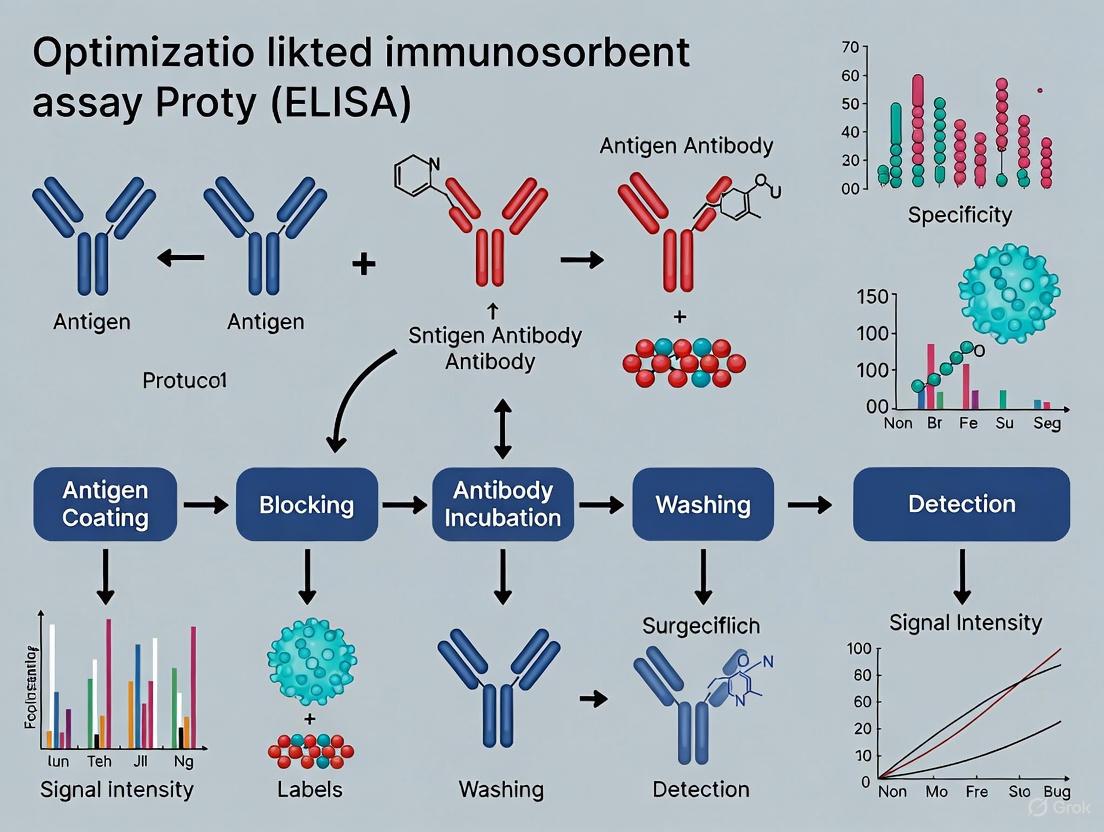
The enzyme-linked immunosorbent assay (ELISA) is a foundational plate-based technique designed for the detection and quantification of soluble substances such as peptides, proteins, antibodies, and hormones [13]. First described by Engvall and Perlmann in 1971, this powerful method enables the analysis of protein samples immobilized in microplate wells using specific antibodies, leveraging the high specificity of antibody-antigen interactions [13] [3]. The fundamental principle of ELISA relies on immobilizing an antigen of interest on a solid surface, complexing it with an antibody linked to a reporter enzyme, and detecting the activity of this enzyme via incubation with a substrate to produce a measurable product [7] [13].
ELISAs are particularly valued for their ability to measure specific analytes within crude preparations, achieved through the immobilization of reagents to the microplate surface, which facilitates easy separation of bound from non-bound material during washing steps [13]. The most common ELISA formats include direct, indirect, sandwich, and competitive assays, with the sandwich format being widely regarded as the most robust and sensitive for its use of two primary antibodies that bind distinct epitopes on the target antigen [7] [13]. This application note deconstructs the complete ELISA workflow, with a particular emphasis on the sandwich ELISA format, providing detailed methodologies and optimization strategies to support researchers in developing robust assays for research and development applications.
Key Stages of the ELISA Workflow
The ELISA procedure consists of several sequential stages, each critical to the assay's overall performance, specificity, and sensitivity. The following workflow diagram illustrates the key stages of a sandwich ELISA, from plate preparation to data analysis.
Stage 1: Plate Coating and Capture
The initial stage of the ELISA workflow involves immobilizing either the antigen or a capture antibody onto the solid surface of a microplate well through passive adsorption driven by hydrophobic interactions between the plastic and non-polar protein residues [13].
- Coating Buffer Selection: The most common coating buffer is 0.2M carbonate/bicarbonate at pH 8.4-9.6, although phosphate-buffered saline (PBS) or Tris-buffered saline (TBS) are sometimes used [14]. The alkaline pH facilitates optimal binding for many proteins.
- Coating Conditions: Proteins are typically diluted to a concentration of 2-10 μg/mL in coating buffer and added to the microplate wells [13]. Plates are then incubated for several hours to overnight at temperatures ranging from 4°C to 37°C [13].
- Alternative Coating Strategies: For oriented antibody binding, plates pre-coated with Protein A, Protein G, or streptavidin can be used, though these should be avoided in sandwich ELISA formats where detection antibodies might also bind to these proteins [14].
Stage 2: Blocking
Following plate coating, the blocking step is crucial to prevent non-specific binding that can cause high background signal. This process involves adding an irrelevant protein or other molecule to cover all remaining unsaturated surface-binding sites of the microplate wells [13].
- Blocking Reagents: Bovine Serum Albumin (BSA) is widely used at concentrations of 1-5% [8]. Normal serums (typically 5% v/v) derived from non-immunized animals are also effective, particularly when diluted in the same host species as the labeled antibody to prevent antibody binding to conserved sequences and Fc-receptors [14].
- Quality Considerations: Commercial BSA preparations may contain contaminating bovine IgG or proteases that can cause background or degrade assay components. Selecting BSA certified as IgG- and protease-free is recommended [14].
- Optimization: Different blocking solutions and concentrations should be tested experimentally. Blocking is typically performed for 1-2 hours at room temperature following coating and prior to sample addition [7] [14].
Stage 3: Sample and Antibody Incubation
This stage encompasses the addition of the sample and the antibodies required for antigen capture and detection.
- Sample Considerations: Samples analyzed by ELISA vary extensively and can include cell lysates, cell culture supernatants, tissue homogenates, and bodily fluids such as serum, urine, and cerebrospinal fluid [14]. Proper sample handling is critical, including storage at -80°C, minimizing freeze-thaw cycles, and keeping samples on ice during use [14].
- Matrix Effects: The sample matrix can significantly affect assay readout. Spike-and-recovery experiments should be performed to assess matrix effects, where a known amount of analyte is added to both the sample matrix and the standard diluent, with ideal results showing little difference between the two conditions [8].
- Antibody Selection: For sandwich ELISAs, a matched antibody pair recognizing different epitopes on the target antigen is required [7]. A common strategy employs a monoclonal antibody for capture and a polyclonal antibody for detection [14].
Stage 4: Detection and Signal Development
Detection in ELISA can be categorized as colorimetric, fluorometric, or chemiluminescent, with the choice dictated by sensitivity requirements, multiplexing needs, and available instrumentation [14].
- Enzyme-Substrate Systems: Horseradish peroxidase (HRP) and alkaline phosphatase (AP) are the most commonly used enzyme labels [13]. The selection of substrate depends on the detection method required and the instrumentation available.
- Detection Methods:
- Colorimetric Detection: Provides a simple, cost-effective readout measured by absorbance. Common substrates include TMB (tetramethylbenzidine) for HRP and pNPP (p-Nitrophenyl Phosphate) for AP [14].
- Fluorometric Detection: Offers greater sensitivity and dynamic range than colorimetric methods, enabling multiplexing capabilities [14].
- Chemiluminescent Detection: Provides exceptional sensitivity and broad dynamic range but requires specialized reagents and instrumentation [14].
Table 1: Common Detection Systems in ELISA
| Detection Method | Enzyme | Common Substrates | Signal Measurement | Sensitivity |
|---|---|---|---|---|
| Colorimetric | HRP | TMB, OPD, ABTS | Absorbance | Moderate |
| Colorimetric | AP | pNPP | Absorbance | Moderate |
| Fluorometric | HRP or AP | Fluorescent substrates | Fluorescence | High |
| Chemiluminescent | HRP | Luminol-based | Luminescence | Very High |
Stage 5: Data Analysis and Quantification
The final stage involves quantifying the target analyte by correlating the assay readout to a standard curve generated on the same microplate using known quantities of the target analyte [14] [15].
- Standard Curve: A dilution series of the target analyte of known concentration is run in parallel with samples to generate a standard curve, which is typically plotted using a 4-parameter logistic regression algorithm [8].
- Calculation: The amount of antigen in unknown samples is calculated by comparing their signal to the standard curve, with appropriate background subtraction and consideration of any dilution factors [8].
- Quality Assessment: The average, standard deviation, and coefficient of variation (%CV) should be determined for replicates to monitor assay performance and data integrity over time [8].
ELISA Optimization and Experimental Protocols
While commercial ELISA kits provide pre-optimized components, researchers developing custom ELISAs must systematically optimize each parameter to ensure robust performance. The following diagram illustrates the optimization workflow, highlighting key parameters requiring systematic evaluation.
Checkerboard Titration Protocol
A checkerboard titration is an efficient experimental approach to optimize multiple ELISA parameters simultaneously, particularly antibody concentrations [7] [8].
- Plate Setup: Prepare different concentrations of the capture antibody in coating buffer and apply equal volumes to the plate in a grid pattern.
- Detection Titration: Similarly, prepare different concentrations of the detection antibody.
- Assay Performance: Proceed with the ELISA protocol using a constant antigen concentration.
- Signal Assessment: Check for strong specific signal versus low background across the different concentration combinations.
- Optimal Concentration Selection: Identify the antibody concentrations that provide the optimal signal-to-noise ratio.
Key Optimization Parameters
Systematic optimization of the following parameters is essential for developing a robust ELISA:
- Antibody Concentrations: The table below provides recommended concentration ranges for coating and detection antibodies based on antibody type and purity [7].
Table 2: Recommended Antibody Concentration Ranges for ELISA Optimization
| Antibody Source | Coating Antibody Concentration | Detection Antibody Concentration |
|---|---|---|
| Polyclonal Serum | 5–15 μg/mL | 1–10 μg/mL |
| Crude Ascites | 5–15 μg/mL | 1–10 μg/mL |
| Affinity-Purified Polyclonal | 1–12 μg/mL | 0.5–5 μg/mL |
| Affinity-Purified Monoclonal | 1–12 μg/mL | 0.5–5 μg/mL |
- Incubation Conditions: Optimization of incubation time and temperature for coating, blocking, and antibody binding steps. While standard protocols often use room temperature incubations of 1-2 hours, some applications may benefit from extended incubations at 4°C [13].
- Wash Conditions: The choice of wash buffer, number of washes, and wash duration significantly impact background signal. Typically, PBS or TBS with 0.05% Tween-20 is used as a wash buffer [8].
- Enzyme Conjugate Concentration: The recommended concentration ranges for enzyme conjugates vary based on the detection system used, as shown in the table below [7].
Table 3: Recommended Enzyme Conjugate Concentrations for ELISA
| Enzyme | Detection System | Recommended Concentration |
|---|---|---|
| HRP | Colorimetric | 20–200 ng/mL |
| HRP | Chemifluorescent | 25–50 ng/mL |
| HRP | Chemiluminescent | 10–100 ng/mL |
| AP | Colorimetric | 100–200 ng/mL |
| AP | Chemiluminescent | 40–200 ng/mL |
Assay Validation Protocols
Comprehensive validation is essential to ensure ELISA accuracy, precision, and reliability for quantitative measurements.
Spike-and-Recovery Experiments:
- Add a known amount of purified analyte to both the sample matrix and the standard diluent.
- Run the ELISA and calculate the recovered concentration for both conditions.
- The percentage recovery should be between 80-120%, with little difference between matrix and diluent [8].
Dilutional Linearity:
- Serially dilute a sample containing a high concentration of the analyte.
- Analyze each dilution and calculate the observed concentration.
- The results should show linearity with the dilution factor, confirming consistent analyte detection across the assay range [8].
Parallelism Testing:
- Serially dilute samples with naturally high analyte concentrations.
- Calculate the concentration of each dilution and determine the coefficient of variation (%CV).
- A high %CV indicates potential matrix effects impacting antibody binding affinity [8].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful ELISA development and implementation requires careful selection of core components. The following table details essential materials and their functions in the ELISA workflow.
Table 4: Essential Research Reagents for ELISA Development
| Reagent/Material | Function | Key Considerations |
|---|---|---|
| Microplate | Solid phase for immobilization | Polystyrene, flat-bottom; clear for colorimetry, black/white for fluorescence/luminescence [14] [13] |
| Coating Antibody | Captures target antigen | Specificity, affinity, concentration (1-15 μg/mL depending on purity) [7] |
| Detection Antibody | Binds captured antigen | Recognizes different epitope than capture antibody; often enzyme-conjugated [7] |
| Blocking Buffer | Prevents non-specific binding | BSA (1-5%) or normal serum (5%); must be protein-rich [14] [13] |
| Coating Buffer | Stabilizes coating protein | Carbonate/bicarbonate (pH 9.4) or PBS (pH 7.4); protein-free [14] |
| Wash Buffer | Removes unbound reagents | PBS or TBS with 0.05% Tween-20 detergent [8] |
| Enzyme Conjugate | Signal generation | HRP or AP conjugated to detection antibody or secondary antibody [7] |
| Substrate | Enzyme converted to detectable product | Colorimetric, fluorogenic, or chemiluminescent based on detection needs [14] |
| Standards | Quantification reference | Known concentrations of pure analyte for standard curve generation [8] |
The ELISA workflow comprises multiple interconnected stages, each requiring careful optimization to achieve maximal assay performance. From initial plate coating and blocking to final data analysis, attention to technical details at each step is essential for developing a robust, sensitive, and reproducible assay. By systematically optimizing critical parameters such as antibody concentrations, incubation conditions, and detection systems through structured approaches like checkerboard titration, researchers can create ELISAs capable of precise protein quantification across diverse applications. The protocols and guidelines presented in this application note provide a framework for researchers to deconstruct and master each element of the ELISA workflow, supporting advancements in biomedical research, drug development, and diagnostic applications.
The Enzyme-Linked Immunosorbent Assay (ELISA) is a powerful, plate-based technique designed for detecting and quantifying soluble substances such as peptides, proteins, antibodies, and hormones [13]. Its fundamental principle relies on the highly specific interaction between an antigen and an antibody, where the antigen is immobilized on a solid surface and complexed with an antibody linked to a reporter enzyme [13]. Detection is accomplished by measuring the activity of this reporter enzyme via incubation with a substrate to produce a measurable product [13]. The performance of an ELISA is critically dependent on the careful selection and optimization of its core components: the solid phase, detection enzymes, substrates, and the reading instrument. Within the context of protocol optimization research, a systematic approach to evaluating these reagents and equipment is paramount for developing assays that are robust, sensitive, and reproducible, thereby providing reliable data for drug development and biomedical research.
Essential Reagents and Their Functions
Solid Phases: Microplates
The solid phase, typically a microplate, serves as the foundational support for the assay, facilitating the immobilization of reactants and the separation of bound from unbound material [13].
- Material and Binding: ELISA plates are usually made of polystyrene and facilitate the passive adsorption of proteins via hydrophobic interactions [13] [16]. This binding is a passive process, and the high protein-binding capacity of the plates is crucial for assay sensitivity.
- Plate Type: For colorimetric detection, clear polystyrene flat-bottom plates are used. For fluorescent or chemiluminescent signals, black or white opaque plates are employed to minimize cross-talk and light reflection [13].
- Selection Criteria: When selecting a plate, key parameters include a high protein-binding capacity (e.g., >400 ng/cm²) and a low coefficient of variation (CV <5%) to ensure well-to-well and plate-to-plate reproducibility [13]. Plates should be visually inspected for imperfections that could cause aberrations in data acquisition [13].
Enzymes and Substrates
The enzyme conjugate and its corresponding substrate form the core of the signal generation system in ELISA. The most commonly used enzyme labels are Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP) [13] [17].
- Horseradish Peroxidase (HRP): A popular choice due to its high specific activity and rapid turnover. Its common substrates include:
- Alkaline Phosphatase (AP): Known for its stability and linear kinetics. A common substrate is pNPP (p-Nitrophenyl Phosphate), which yields a yellow colorimetric product measurable at 405-410 nm [17].
The choice between a colorimetric, chemiluminescent, or fluorescent substrate depends on the required assay sensitivity and the available detection instrumentation [13]. Chemiluminescent substrates generally offer the highest sensitivity [7].
Key Research Reagent Solutions
A successful ELISA requires a suite of carefully formulated buffers and reagents. The table below summarizes the essential materials and their functions.
Table 1: Essential Reagents for ELISA Development and Optimization
| Reagent Category | Specific Examples | Function in the Assay |
|---|---|---|
| Coating Buffer | Carbonate-bicarbonate buffer (pH 9.4), PBS (pH 7.4) [13] | Provides optimal pH and ionic conditions for passive adsorption of antigen or capture antibody to the plate. |
| Blocking Buffer | PBS with 0.1% BSA and 0.05% Tween 20 [16] | Saturates all remaining protein-binding sites on the plate to minimize non-specific binding and reduce background signal. |
| Wash Buffer | PBS containing 0.05% Tween 20 [16] | Removes unbound reagents and weakly associated molecules during the washing steps; Tween 20 blocks newly exposed sites. |
| Sample Diluent | Incubation buffer (for cell culture supernatants), specialized ELISA diluent (for serum/plasma) [16] | Dilutes samples and standards in a matrix that mimics the sample to prevent interference and maintain antibody-antigen binding. |
| Detection Antibodies | Biotinylated primary antibody, Enzyme-conjugated secondary antibody, Streptavidin-HRP [16] [7] | Binds specifically to the captured antigen (directly or indirectly) and carries the enzyme label for signal generation. |
| Enzyme Conjugate | Streptavidin-HRP, Anti-species IgG-HRP [7] | Provides the catalytic enzyme (e.g., HRP, AP) that converts the substrate into a detectable signal. |
| Stop Solution | 0.16 M Sulfuric Acid [7] | Halts the enzyme-substrate reaction abruptly at a defined endpoint, stabilizing the signal for measurement. |
Experimental Protocols for Optimization
Protocol optimization is a systematic process to ensure the assay delivers strong, specific signals with low background. The following methodologies are critical for this process.
Checkerboard Titration for Reagent Optimization
A checkerboard titration is an efficient experimental design used to optimize the concentrations of two key reagents simultaneously, such as the capture and detection antibodies [7].
Detailed Protocol:
- Prepare Capture Antibody Dilutions: Dilute the capture antibody in coating buffer across a range of concentrations (e.g., 1-15 µg/mL, depending on source and purity) [7]. Dispense different concentrations in rows of the microplate.
- Coat and Block: Incubate the plate overnight at 4°C, then wash and block the plate using a standard blocking buffer.
- Add Antigen: Add a fixed, known concentration of the target antigen (a purified standard) to all wells.
- Prepare Detection Antibody Dilutions: Dilute the detection antibody in standard diluent across a range of concentrations (e.g., 0.5-10 µg/mL) [7]. Dispense different concentrations in columns of the microplate.
- Complete the Assay: Incubate, wash, and then add the enzyme conjugate (if the detection antibody is not directly conjugated) and substrate following standard procedures.
- Data Analysis: Measure the signal. The optimal combination is the lowest concentration of both antibodies that yields the highest signal-to-noise ratio (strong signal with low background) [7].
Protocol for Assay Validation: Spike-and-Recovery
The spike-and-recovery experiment determines if components in the sample matrix (e.g., serum, cell culture media) interfere with the detection of the target analyte [9].
Detailed Protocol:
- Prepare Spiked Samples: Spike a known concentration of the purified standard protein into both the sample matrix of interest and a standard diluent (e.g., assay buffer). Prepare multiple dilutions.
- Prepare Control Samples: Include a non-spiked sample of the matrix and the diluent as background controls.
- Run the ELISA: Analyze all samples using the optimized ELISA protocol.
- Calculate Percent Recovery: For each spiked sample, use the formula:
- % Recovery = (Measured concentration in spiked matrix / Measured concentration in spiked diluent) × 100
- Interpretation: Recovery values between 80% and 120% are generally acceptable, indicating minimal matrix interference. If recovery is outside this range, the sample matrix may require further dilution, or the standard curve may need to be prepared in the same matrix [9].
Data Analysis and Standard Curve Generation
Accurate quantification in a quantitative ELISA requires the generation of a standard curve from which the concentration of unknown samples is extrapolated [9] [18].
Detailed Protocol:
- Serial Dilution: Perform a serial dilution of the purified antigen standard in the chosen diluent to generate a range of known concentrations.
- Run Standards and Samples: Analyze the standard dilutions and unknown samples in duplicate or triplicate.
- Plot and Curve Fit: Plot the mean absorbance (y-axis) against the protein concentration (x-axis). Use appropriate curve-fitting models:
- Calculate Unknowns: For each unknown sample, find its average absorbance on the y-axis, extend a horizontal line to the standard curve, and then a vertical line down to the x-axis to read the corresponding concentration. Multiply by the dilution factor if the sample was diluted [9] [18].
- Assess Precision: Calculate the coefficient of variation (CV) for sample duplicates. A CV of ≤20% is typically acceptable, with larger values indicating inconsistency or error [9] [18].
Table 2: Recommended Concentrations for ELISA Optimization
| Reagent | Source | Recommended Optimization Range | Key Consideration |
|---|---|---|---|
| Coating Antibody | Affinity-purified polyclonal or monoclonal | 1 - 12 µg/mL [7] | Use purified antibodies for optimal signal-to-noise ratio. |
| Detection Antibody | Affinity-purified polyclonal or monoclonal | 0.5 - 5 µg/mL [7] | Must be specific for the primary antibody (in indirect/detection). |
| HRP Conjugate | Colorimetric System | 20 - 200 ng/mL [7] | Concentration must be compatible with the substrate's range. |
| AP Conjugate | Colorimetric System | 100 - 200 ng/mL [7] | Known for stable, linear reaction kinetics. |
Workflow and Relationships
The following diagram illustrates the logical workflow and decision-making process involved in optimizing and executing a sandwich ELISA, from initial setup to data analysis.
ELISA Optimization and Execution Workflow
The relationships between core ELISA reagents and the resulting signal are fundamental to the assay's function. The diagram below depicts this critical signaling pathway.
ELISA Signal Generation Pathway
Advanced Methodologies and Practical Application Strategies
The Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technology for detecting and quantifying target analytes within complex biological samples. The selection of an appropriate assay format—direct, indirect, sandwich, or competitive—is a critical determinant of experimental success, impacting sensitivity, specificity, workflow efficiency, and data accuracy. This application note provides a comprehensive comparative analysis of these core ELISA formats, underpinned by current optimization research. We present structured quantitative data, detailed experimental protocols, and best-practice reagent specifications to guide researchers and drug development professionals in selecting and optimizing the most suitable ELISA configuration for their specific application needs, thereby enhancing the reliability and reproducibility of data within rigorous scientific and regulatory frameworks.
The fundamental principle of ELISA involves the immobilization of an antigen (Ag) or antibody (Ab) onto a solid phase (typically a polystyrene microplate), followed by a series of binding and amplification steps that ultimately generate a measurable signal proportional to the analyte concentration [13] [4]. The specificity of the assay is conferred by robust antigen-antibody interactions, while sensitivity is achieved by conjugating enzymes, such as Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP), to antibodies or antigens. These enzymes catalyze the conversion of a substrate into a colored, fluorescent, or luminescent product that can be quantified spectrophotometrically [19] [13]. The versatility of ELISA is demonstrated by its compatibility with diverse sample matrices, including serum, plasma, cell culture supernatants, saliva, and tissue lysates [19]. The evolution of the technique from its inception in the 1970s has led to the development of multiple formats, each with distinct advantages and limitations, making format selection a primary consideration in assay design [4].
Comparative Analysis of ELISA Formats
The four principal ELISA formats—direct, indirect, sandwich, and competitive—differ in their configuration of antibody-antigen interactions, which directly influences their application-specific performance. The table below provides a structured comparison of these core characteristics.
Table 1: Comprehensive Comparison of Core ELISA Formats
| Format | Key Principle | Best Applications | Advantages | Disadvantages |
|---|---|---|---|---|
| Direct ELISA [19] [20] | A labeled primary antibody binds directly to the immobilized antigen. | Assessing antibody affinity and specificity; blocking/inhibitory studies [19]. | Fast, simple protocol; minimal steps; reduced cross-reactivity risk [19] [13]. | Lower sensitivity; potential for high background; limited antibody options [19] [20]. |
| Indirect ELISA [19] [13] | An unlabeled primary antibody binds the antigen, and is detected by a labeled secondary antibody. | Detecting endogenous antibodies (e.g., serological testing) [19] [21]. | High sensitivity due to signal amplification; versatile; wide range of available secondary antibodies [19] [20]. | Risk of cross-reactivity from secondary antibody; longer protocol [19] [13]. |
| Sandwich ELISA [19] [7] | The antigen is captured between a surface-bound antibody and a detection antibody. | Quantifying specific antigens in complex samples (e.g., cytokines, biomarkers) [19] [21]. | High sensitivity and specificity; compatible with complex samples; no sample purification required [19] [22]. | Requires two matched antibodies; longer development time; challenging to optimize [19] [20]. |
| Competitive ELISA [19] [20] | Sample antigen and labeled antigen compete for a limited number of antibody binding sites. | Quantifying small molecules (e.g., hormones, drugs, contaminants) [19] [21]. | Ideal for small antigens; robust with complex samples; flexible format [19] [20]. | Lower sensitivity; signal is inversely proportional to analyte; requires optimization [19] [20]. |
The strategic selection among these formats hinges on the molecular characteristics of the analyte (e.g., size, availability of epitopes), the required sensitivity and specificity, and the available reagents. The trend in next-generation ELISA (ELISA 2.0) is toward digital detection, single-molecule sensing (e.g., digital ELISA), and multiplexing to overcome the limitations of traditional formats, offering ultra-sensitive, high-throughput analysis crucial for advanced diagnostics and biopharmaceutical quality control [23].
Experimental Protocols and Workflows
A successful ELISA requires a meticulously optimized, step-by-step protocol. The following sections detail the standard workflows for the two most common formats: Sandwich and Competitive ELISA.
Detailed Protocol: Sandwich ELISA
The sandwich ELISA is the preferred format for quantifying specific proteins and biomarkers due to its superior specificity [7] [22].
Workflow Overview:
Sandwich ELISA Workflow
Step-by-Step Methodology:
Plate Coating: Dilute the capture antibody in a coating buffer (e.g., carbonate-bicarbonate buffer, pH 9.4 or phosphate-buffered saline (PBS), pH 7.4) to a concentration typically between 1–15 µg/mL, with affinity-purified antibodies often optimal at 1–12 µg/mL [7] [13]. Dispense 100 µL/well into a 96-well microplate. Seal the plate and incubate for a minimum of 2 hours at room temperature or overnight at 4°C.
Blocking: Aspirate the coating solution. Wash the plate three times with 300 µL/well of wash buffer (e.g., PBS containing 0.05% Tween 20, PBST). Add 200–300 µL/well of blocking buffer (e.g., 1–5% BSA or 5% non-fat dry milk in PBST) to cover all unsaturated binding sites. Incubate for 1–2 hours at room temperature [7] [8].
Sample and Standard Incubation: Aspirate the blocking buffer and wash the plate 3 times. Prepare serial dilutions of the protein standard in a diluent that closely matches the sample matrix (e.g., cell culture medium, assay buffer) [7]. Add 100 µL/well of standards, controls, and test samples. Incubate for 1–2 hours at room temperature to allow antigen capture.
Detection Antibody Incubation: Wash the plate 3–5 times to remove unbound antigen. Add 100 µL/well of the biotinylated or enzyme-conjugated detection antibody, diluted in diluent to a concentration of 0.5–10 µg/mL [7]. Incubate for 1–2 hours at room temperature.
Enzyme Conjugate Incubation: Wash the plate as before. If using a biotinylated detection antibody, add 100 µL/well of Streptavidin-HRP conjugate diluted in diluent to the recommended concentration (e.g., 20–200 ng/mL for HRP colorimetric systems) [7]. Incubate for 30–60 minutes at room temperature, protected from light.
Signal Development: Perform a final wash step (5 times is recommended). Add 100 µL/well of a suitable substrate solution (e.g., TMB for HRP). Incubate for 5–30 minutes at room temperature, protected from light, until optimal color development is achieved.
Signal Detection and Analysis: Stop the enzyme-substrate reaction by adding 50–100 µL/well of stop solution (e.g., 0.16M sulfuric acid for TMB) [7]. Immediately measure the absorbance at the appropriate wavelength (e.g., 450 nm for TMB) using a microplate reader. Generate a standard curve by plotting the mean absorbance versus the standard concentration using a 4-parameter logistic (4-PL) regression model to interpolate sample concentrations [8].
Detailed Protocol: Competitive ELISA
Competitive ELISA is primarily used for measuring small molecules and hormones that are too small to be bound by two antibodies simultaneously [19] [20].
Workflow Overview:
Competitive ELISA Workflow
Step-by-Step Methodology:
Plate Coating: Coat the microplate with a known quantity of purified antigen (typically 2–10 µg/mL in coating buffer) [13] [20]. Incubate and wash as described in the sandwich protocol.
Blocking: Block the plate with an appropriate protein-based blocking buffer to prevent non-specific binding.
Competitive Incubation: Pre-incubate a constant, limited concentration of the enzyme-labeled antibody with serially diluted samples or standards containing the target antigen. Then, add this mixture to the antigen-coated plate. Alternatively, the labeled antibody can be added directly to the wells simultaneously with the sample. The key principle is that the antigen in the sample and the immobilized antigen compete for binding sites on the labeled antibody [19] [20]. Incubate for 1–2 hours at room temperature.
Wash: Wash the plate thoroughly to remove any unbound labeled antibody. The amount of antibody bound to the plate is inversely proportional to the concentration of antigen in the sample.
Signal Development and Detection: Add substrate solution to develop the signal. Stop the reaction and read the absorbance. Higher analyte concentration in the sample results in less antibody bound to the plate and, consequently, a lower signal [19].
The Scientist's Toolkit: Essential Reagents and Materials
The consistency and performance of an ELISA are dependent on the quality and optimization of its core components. The following table outlines the essential reagents required.
Table 2: Essential Research Reagent Solutions for ELISA Development
| Reagent/Material | Function | Key Specifications & Best Practices |
|---|---|---|
| Microplate [13] | Solid phase for immobilization of capture antibody or antigen. | Use high-protein-binding polystyrene plates (clear for colorimetric, white/black for chemiluminescent/fluorescent); ensure low well-to-well variation (CV <5%). |
| Coating Antibody [7] | Binds and immobilizes the target antigen from the sample. | Use affinity-purified antibodies; optimize concentration (1–12 µg/mL for purified monoclonals/polyclonals); select an antibody with high affinity and specificity. |
| Detection Antibody [7] [22] | Binds to a different epitope on the captured antigen. | Must be a matched pair with the capture antibody; can be biotinylated or directly conjugated to an enzyme (e.g., HRP); optimize concentration (0.5–5 µg/mL for purified antibodies). |
| Blocking Buffer [7] [8] | Covers unsaturated binding sites to minimize non-specific background signal. | Common agents: 1–5% BSA, 5% non-fat dry milk, or serum in PBST; requires empirical testing for optimal performance with specific antibody-antigen pairs. |
| Enzyme Conjugate [7] | Catalyzes the substrate to generate a detectable signal. | Common enzymes: HRP and Alkaline Phosphatase (AP). Optimize concentration (e.g., 20–200 ng/mL for HRP colorimetric). Streptavidin-HRP is used with biotinylated detection antibodies. |
| Substrate [13] | Converted by the enzyme into a measurable product. | TMB (colorimetric, HRP) is common. For higher sensitivity, use chemiluminescent substrates. Ensure compatibility with the enzyme and detection instrument. |
| Wash Buffer [4] | Removes unbound reagents and reduces background. | Typically PBS or Tris-based buffer with a surfactant (e.g., 0.05% Tween 20). Sufficient wash volume and cycles (3-5x) are critical for low background. |
Critical Protocol Optimization and Validation
Assay development does not end with establishing a workflow; rigorous optimization and validation are imperative for generating reliable, reproducible data, particularly in a drug development context.
Optimization via Checkerboard Titration
A checkerboard titration is the most efficient method for simultaneously optimizing the concentrations of the capture and detection antibodies [7] [8]. Prepare a series of dilutions of the capture antibody and coat them across the plate in rows. Similarly, prepare dilutions of the detection antibody and add them down the columns. After running the assay, analyze the signal-to-background ratio for each well. The optimal condition is the combination of antibody concentrations that yields the strongest specific signal with the lowest background, providing the widest assay dynamic range [7].
Essential Validation Experiments
- Spike and Recovery: Assess the impact of the sample matrix by spiking a known amount of the analyte into the sample matrix and a reference diluent. Calculate the percentage recovery; ideal recovery is 80–120%, indicating minimal matrix interference [8].
- Dilutional Linearity: Serially dilute a sample with a high endogenous concentration of the analyte. The measured concentrations, when corrected for dilution, should be constant. A loss of linearity indicates matrix effects or assay hook effect, necessitating further optimization of the sample diluent [8].
- Parallelism: Compare the standard curve diluted in assay buffer to the standard curve diluted in a matrix similar to the sample (e.g., normal serum). The curves should be parallel, confirming that the antibody affinity is similar for the standard and the endogenous analyte [8].
The strategic selection and meticulous optimization of an ELISA format are foundational to successful research and assay development. The direct and indirect formats offer simplicity and speed for specific applications, while the sandwich ELISA provides unparalleled specificity for quantifying proteins in complex matrices. The competitive format is indispensable for the analysis of small molecules. The ongoing evolution of ELISA, through advancements in detection chemistry, multiplexing, and automation (ELISA 2.0), continues to expand its utility in biomarker discovery, diagnostics, and biopharmaceutical quality control [23]. By adhering to the detailed protocols, optimization strategies, and validation practices outlined in this document, scientists can ensure their ELISA methods are robust, sensitive, and fit-for-purpose.
The Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technique for the quantitative detection of proteins, peptides, antibodies, and hormones in biological samples [4]. Its robustness, however, is critically dependent on meticulous optimization of key procedural steps to ensure high sensitivity, specificity, and reproducibility. Within the broader context of ELISA protocol optimization research, this application note delineates three fundamental checkpoints: antibody coating, blocking buffers, and sample diluents. Failures at any of these stages can manifest as excessive background noise, diminished signal strength, poor precision, or inaccurate quantification, ultimately compromising data integrity [14] [7]. This guide provides detailed methodologies and data-driven recommendations to empower researchers in systematically optimizing these parameters for robust assay performance.
Antibody Coating Optimization
The initial immobilization of the capture antibody onto the microplate surface is the foundation of a sandwich ELISA. Passive adsorption via hydrophobic interactions is the most common method, and its efficiency dictates the assay's ultimate capacity [14].
Coating Buffer Composition and pH
The choice of coating buffer is paramount for stabilizing the biomolecule and facilitating its binding to the polystyrene plate. A summary of common coating buffers is provided in Table 1.
Table 1: Common Coating Buffers for ELISA
| Buffer Type | Typical Composition | Optimal pH Range | Key Considerations |
|---|---|---|---|
| Carbonate/Bicarbonate | 0.2 M carbonate/bicarbonate [14] | 8.4 - 9.6 [14] | Most common; high pH enhances hydrophobic binding for many proteins. pH choice depends on protein isoelectric point [14]. |
| Phosphate-Buffered Saline (PBS) | 10 mM phosphate, 137 mM NaCl, 2.7 mM KCl [24] | 7.2 - 7.4 [24] | Physiological pH; suitable for some antibodies and antigens sensitive to alkaline conditions. |
| Tris-Buffered Saline (TBS) | 25 mM Tris, 150 mM NaCl [24] | 7.2 - 7.4 [24] | Alternative to PBS; note that phosphate can interfere with alkaline phosphatase (AP)-based detection systems [25]. |
Critical Consideration: The coating buffer must be protein-free to prevent competitive binding of extraneous proteins to the plate, which becomes a significant source of non-specific background [14].
Experimental Protocol: Checkerboard Titration for Coating and Detection Antibodies
A checkerboard titration is the most efficient method to simultaneously optimize the concentrations of both the capture and detection antibodies [7] [8].
Materials:
- Purified capture antibody
- Coating buffer (e.g., Carbonate-Bicarbonate, pH 9.6)
- Blocking buffer (e.g., 5% BSA or commercial blocker)
- Antigen standard
- Detection antibody (biotinylated or directly conjugated)
- Wash buffer (e.g., PBS or TBS with 0.05% Tween-20)
- Enzyme conjugate (e.g., Streptavidin-HRP)
- Substrate and Stop solution
Method:
- Prepare Capture Antibody Dilutions: Dilute the capture antibody in coating buffer across a range of concentrations (e.g., 1-15 µg/mL for affinity-purified antibodies) [7].
- Coat Plate: Add different concentrations of the capture antibody to the microplate wells in a grid pattern. Incubate overnight at 4°C or for 1-3 hours at 37°C.
- Wash and Block: Wash the plate 2-3 times with wash buffer. Add a sufficient volume of blocking buffer and incubate for 1-2 hours at room temperature.
- Add Antigen: Wash the plate. Add a fixed, known concentration of the antigen standard to all wells.
- Prepare Detection Antibody Dilutions: Dilute the detection antibody in blocking or assay buffer across a range of concentrations.
- Detect: Wash the plate. Add different concentrations of the detection antibody to the plate in an orthogonal grid pattern relative to the capture antibody. Incubate and wash.
- Complete Assay: Add the enzyme conjugate, incubate, wash, and then add substrate. Finally, stop the reaction and read the absorbance.
Data Analysis: The optimal condition is the pair of the lowest antibody concentrations that yields the highest signal-to-noise ratio (i.e., strong positive signal with low background in negative controls) [7].
The following diagram illustrates the logical workflow and key decision points for optimizing the three critical checkpoints described in this application note.
Blocking Buffer Selection
After coating, any remaining protein-binding sites on the polystyrene plate must be blocked to prevent non-specific adsorption of assay components, a major contributor to high background signal [14] [25].
Types of Blocking Buffers
No single blocking agent is ideal for every application, as each antibody-antigen pair has unique characteristics [25]. The choice often involves a trade-off between blocking efficiency and potential interference with antigen-antibody binding. Key options are summarized in Table 2.
Table 2: Common Blocking Buffers for ELISA
| Blocking Agent | Typical Concentration | Benefits | Considerations and Potential Interferences |
|---|---|---|---|
| Bovine Serum Albumin (BSA) | 1-5% [25] | Highly purified, inexpensive, compatible with biotin-streptavidin systems and phosphoprotein detection [25] [24]. | Generally a weaker blocker than others, which can result in more non-specific binding. Commercial preparations may contain contaminating bovine IgG and proteases [14]. |
| Non-Fat Dry Milk | 2-5% [25] | Inexpensive and effective; contains multiple protein types. | Contains biotin and phosphoproteins, which can interfere with streptavidin-biotin detection and phospho-protein analysis [25]. May mask some antigens. |
| Normal Sera | 2-5% v/v [14] [26] | Excellent for preventing antibody binding to conserved sequences and Fc-receptors. Best if from the same species as the labeled antibody [14]. | Risk of falsely elevated signal if the primary antibody binds to a serum protein [14]. Can be more variable. |
| Casein | 1-2% [25] [24] | Single purified protein; fewer cross-reactions than milk. Good high-performance replacement for milk [24]. | More expensive than milk or BSA. |
| Protein-Free Blockers | Ready-to-use [24] [26] | Contains no animal proteins; ideal for eliminating cross-reactivity from antibodies reacting with blocking proteins. Minimizes background in sensitive assays [24] [26]. | Can be more costly. Performance is formulation-dependent. |
Experimental Protocol: Comparing Blocking Buffer Efficacy
Materials:
- Coated and washed microplate
- Candidate blocking buffers (e.g., 5% BSA, 5% Non-Fat Milk, 1% Casein, Commercial Protein-Free blocker)
- Antigen standard
- Detection system (antibodies, conjugate, substrate)
Method:
- Divide Plate: After coating and washing, divide the plate into sections for each blocking buffer to be tested.
- Block: Add the different blocking buffers to their respective sections. Incubate for 1-2 hours at room temperature.
- Run Assay: Wash the plate. Proceed with the standard ELISA protocol, using the same concentrations of antigen, detection antibody, and conjugate across all sections.
- Measure: Read the final absorbance.
Data Analysis: The optimal blocking buffer is the one that yields the highest signal in positive control wells (containing antigen) and the lowest signal in negative control wells (no antigen), resulting in the best signal-to-noise ratio [25]. High background across all blockers may indicate insufficient blocking time or concentration, while signal loss may suggest the blocker is interfering with antibody-antigen binding.
Sample Diluent and Matrix Effects
The sample matrix (e.g., serum, plasma, cell culture supernatant) can profoundly influence assay performance through matrix effects, which can artificially suppress or enhance the signal [14] [8].
Key Validation Experiments
Spike and Recovery: This experiment assesses the extent to which the sample matrix affects the accurate quantification of the analyte [14] [8].
- Method: A known concentration of the purified analyte (the "spike") is added to a sample matrix and to a well-characterized diluent (e.g., the standard diluent buffer). Both are run in the ELISA, and the measured concentration is compared to the expected concentration.
- Calculation: % Recovery = (Measured concentration in spiked sample / Expected concentration) × 100.
- Interpretation: Recoveries of 80-120% are generally acceptable. Poor recovery indicates significant matrix interference, necessitating sample dilution or a change in standard diluent to one that more closely matches the sample matrix [8].
Dilutional Linearity: This test determines whether the assay can accurately measure the analyte across different dilutions and identifies the linear range of the assay [8].
- Method: A sample with a high endogenous concentration of the analyte (or a spiked sample) is serially diluted in the chosen sample diluent. Each dilution is run in the ELISA.
- Data Analysis: The observed concentration for each dilution is plotted against the dilution factor. The data should fall along a straight line. A lack of linearity (e.g., curves upward or downward) suggests matrix effects that are not corrected by dilution, requiring further optimization of the diluent [8].
The Scientist's Toolkit: Essential Reagents for ELISA Optimization
A well-stocked laboratory is crucial for efficient ELISA development and troubleshooting. Table 3 lists key reagent solutions and their functions.
Table 3: Essential Research Reagent Solutions for ELISA Development
| Reagent Category | Specific Examples | Primary Function |
|---|---|---|
| Coating Buffers | Carbonate-Bicarbonate Buffer (pH 9.4-9.6), Phosphate-Buffered Saline (PBS, pH 7.4) [14] [24] | To immobilize capture antibodies or antigens onto the microplate surface via passive adsorption. |
| Blocking Buffers | BSA (1-5%), Non-Fat Dry Milk (2-5%), Normal Sera (2-5%), Casein, Commercial Protein-Free Blockers [25] [24] [26] | To cover any remaining protein-binding sites on the plate to minimize non-specific binding and reduce background. |
| Wash Buffers | PBS with 0.05% Tween-20 (PBS-T), TBS with 0.05% Tween-20 (TBS-T) [24] | To remove unbound reagents and proteins between assay steps, reducing background signal. |
| Sample / Assay Diluents | Commercial ELISA Assay Buffer, PBS or TBS with a low concentration of protein (e.g., 1% BSA) [24] [27] | To dilute samples, standards, and detection antibodies while maintaining protein stability and minimizing matrix effects. |
| Detection Enzymes & Substrates | Horseradish Peroxidase (HRP)/TMB, Alkaline Phosphatase (AP)/pNPP [14] | To generate a measurable signal (colorimetric, chemiluminescent, fluorescent) proportional to the amount of captured analyte. |
Systematic optimization of antibody coating, blocking buffers, and sample diluents is non-negotiable for developing a robust and reliable ELISA. These three checkpoints are deeply interconnected; for instance, an inefficient block will lead to high background regardless of antibody quality, and an inappropriate sample diluent can cause inaccurate quantification even in a perfectly coated plate. By employing the structured experimental protocols outlined herein—particularly checkerboard titrations, comparative blocking studies, and spike/recovery assays—researchers can directly and quantitatively assess the impact of each variable. This data-driven approach moves optimization beyond trial-and-error, enabling the development of ELISA methods with the high sensitivity, specificity, and reproducibility required for critical research and drug development applications.
The Enzyme-Linked Immunosorbent Assay (ELISA) represents a cornerstone technology in immunology, diagnostics, and biomedical research, but its accuracy and reliability are profoundly influenced by meticulous optimization procedures. Systematic optimization addresses numerous challenges inherent in immunoassay development, including antigen-antibody interaction dynamics, matrix interference effects, and signal amplification variability. Without rigorous optimization, ELISA results can be compromised by false positives, high background noise, poor reproducibility, and inadequate sensitivity [28] [29]. Among the various methodological approaches available, Checkerboard Titration and the Taguchi Method have emerged as powerful, efficient strategies for navigating the multidimensional parameter space of ELISA development. These techniques enable researchers to methodically identify optimal assay conditions while conserving valuable reagents and time [8] [30]. This application note provides detailed protocols for implementing these systematic optimization techniques within the context of ELISA development, complete with structured data presentation, experimental workflows, and reagent specifications tailored for research scientists and drug development professionals.
Checkerboard Titration for ELISA Optimization
Principle and Application
Checkerboard titration is a fundamental experimental design that allows for the simultaneous optimization of two critical ELISA parameters, most commonly the concentrations of capture and detection antibodies. This method employs a grid-based approach where one parameter is varied across the rows of a microtiter plate while the second parameter is varied down the columns, creating a matrix of different concentration combinations [8] [31]. All other assay conditions are maintained constant during this process. The primary objective is to identify the specific combination that yields the maximal assay window—defined as the difference between the positive signal (full signal) and background (nonspecific binding) [8]. This technique is particularly valuable during the initial stages of assay development for determining the optimal working concentrations of matched antibody pairs in sandwich ELISA formats, or for optimizing antigen and antibody concentrations in other formats [30].
Experimental Protocol
Materials Required:
- Coating antibody (capture antibody) at known concentration
- Detection antibody (if using sandwich ELISA)
- Target antigen (standard) of known concentration
- Appropriate microtiter plates (e.g., high protein binding)
- Coating buffer (e.g., carbonate/bicarbonate buffer, pH 9.6)
- Blocking buffer (e.g., BSA, casein, or commercial blockers)
- Wash buffer (e.g., PBS or Tris with Tween 20)
- Detection reagents (enzyme-conjugated secondary antibody if applicable)
- Substrate solution (appropriate for enzyme conjugate)
- Stop solution (if required)
- Plate reader capable of measuring the appropriate signal (absorbance, fluorescence, or luminescence)
Procedure:
- Prepare Coating Antibody Dilutions: Create a series of dilutions for the capture antibody in coating buffer. The recommended starting range for affinity-purified antibodies is typically 1-12 µg/mL, while for unpurified antisera or ascites, 5-15 µg/mL is appropriate [30] [31].
- Plate Coating: Apply equal volumes of each capture antibody dilution across the rows of the microtiter plate (e.g., from row A to row H). Incubate according to standard coating conditions (typically overnight at 4°C or 1-2 hours at 37°C).
- Washing: Wash the plate multiple times (typically 3-5 washes) with wash buffer to remove unbound antibody.
- Blocking: Add an appropriate blocking solution to all wells to cover any remaining protein-binding sites. Incubate for 1-2 hours at room temperature or following established protocols.
- Prepare Detection Antibody Dilutions: During the blocking step, prepare a series of dilutions for the detection antibody. The recommended range for affinity-purified detection antibodies is typically 0.5-5 µg/mL [30] [31].
- Antigen Addition: After washing the blocked plate, add a fixed, optimal concentration of target antigen to all wells, or if optimizing antigen concentration simultaneously, titrate the antigen across a different dimension of the plate.
- Detection Antibody Incubation: Apply the detection antibody dilutions down the columns of the plate (e.g., from column 1 to column 12). If using a direct detection system, this antibody will be enzyme-conjugated; if using an indirect system, this will be followed by an enzyme-conjugated secondary antibody.
- Signal Development: After appropriate incubation and washing, add substrate solution to all wells and incubate for a fixed, optimal time period.
- Signal Measurement: Stop the reaction if necessary, and measure the signal using a plate reader with appropriate filters for the detection system.
Data Analysis:
- For each well, calculate the signal-to-noise ratio by dividing the signal from the test well by the signal from negative control wells (wells without antigen or with control samples known to lack the target).
- Identify the combination of capture and detection antibody concentrations that produces the highest signal-to-noise ratio while maintaining a saturating signal level for the expected analyte concentration range.
- Confirm selected conditions by testing with known positive and negative samples across the expected concentration range of the target analyte.
Checkerboard titration workflow for simultaneous optimization of two ELISA parameters, typically capture and detection antibody concentrations.
Expected Outcomes and Interpretation
A successfully executed checkerboard titration will generate a matrix of signal intensities corresponding to different combinations of the two optimized parameters. The ideal outcome is identification of a "sweet spot" where further increases in either antibody concentration do not significantly improve the signal-to-noise ratio, indicating optimal reagent usage without unnecessary consumption [8] [30]. This optimal combination should be validated using control samples with known analyte concentrations to ensure the assay maintains linearity across the expected working range. Researchers should note that the optimal concentrations identified through checkerboard titration may need adjustment when other parameters (e.g., incubation times, temperatures, or buffer compositions) are subsequently modified, necessitating potential iterative optimization cycles [28].
Taguchi Method for Multifactorial Optimization
Principle and Application
The Taguchi Method represents a statistically powerful experimental design approach for optimizing multiple parameters simultaneously with a minimal number of experimental runs. This method employs specially constructed arrays (orthogonal arrays) that allow researchers to efficiently evaluate the individual effects of multiple factors and their potential interactions [28] [32]. Unlike traditional one-variable-at-a-time approaches, the Taguchi Method recognizes that optimal performance often occurs at specific combinations of factor levels that might not be discovered when varying parameters independently [32]. In ELISA development, this method is particularly valuable when numerous parameters require optimization, including antibody concentrations, incubation times, blocking conditions, buffer compositions, and detection system variables [28]. The core outcome metric in Taguchi experiments is the signal-to-noise (S/N) ratio, which simultaneously measures both the magnitude of the response (signal) and its variability (noise), with the explicit goal of maximizing this ratio to achieve robust, reproducible assay performance [32].
Experimental Protocol
Materials Required:
- All standard ELISA reagents as previously listed
- Software for experimental design and statistical analysis (optional)
Procedure:
- Parameter Selection: Identify the critical factors to be optimized (e.g., coating antibody concentration, detection antibody concentration, blocking buffer type, incubation time, temperature). Typically, 4-7 key factors are selected for initial optimization.
- Level Assignment: For each factor, define 2-4 potential levels (specific values or conditions) to be tested. For continuous variables like concentration, select levels that span a reasonable range based on preliminary experiments or literature values.
- Orthogonal Array Selection: Choose an appropriate orthogonal array that can accommodate the number of factors and levels being tested. For example, an L8 array can handle up to 7 factors at 2 levels each with only 8 experimental runs.
- Experimental Execution: Perform ELISA according to the conditions specified for each run in the orthogonal array. Include appropriate controls in each experimental run.
- Signal-to-Noise Ratio Calculation: For each experimental run, calculate the S/N ratio using the appropriate formula based on the experimental goal. For ELISA optimization where maximization of signal with minimal background is desired, the "larger-is-better" S/N ratio is typically used: S/N = -10 × log₁₀(Σ(1/y²)/n), where y represents the response (signal-to-background ratio) for each replication, and n is the number of replications.
- Factor Effect Analysis: Calculate the average S/N ratio for each factor at each level to determine the optimal combination of factor levels.
- Confirmation Experiment: Conduct a confirmation experiment using the predicted optimal conditions to verify improvement in assay performance.
Taguchi method workflow for multifactorial optimization of ELISA parameters using orthogonal arrays and signal-to-noise ratio analysis.
Expected Outcomes and Interpretation
Successful implementation of the Taguchi Method typically results in identification of a factor combination that significantly improves the signal-to-noise ratio compared to baseline conditions. In one documented application, researchers using the Taguchi Method to optimize an immunodetection system achieved an increase in S/N ratio from -12.89 dB to -10.91 dB, corresponding to a 33.1% reduction in quality loss [32]. The analysis generates clear data on the relative contribution of each factor to the overall assay performance, allowing researchers to focus control efforts on the most influential parameters. The confirmation experiment validates that the predicted optimal conditions indeed produce the expected performance improvement. This method not only identifies optimal conditions but also provides insights into the robustness of the assay by revealing how different factors influence variability in results [28] [32].
Research Reagent Solutions
The following table details essential reagents and materials required for implementing systematic ELISA optimization protocols:
Table 1: Essential Research Reagents for ELISA Optimization
| Reagent Category | Specific Examples | Function in ELISA | Optimization Considerations |
|---|---|---|---|
| Antibodies | Capture antibody, Detection antibody, Enzyme-conjugated secondary antibody | Specific binding to target analyte; signal generation | Concentration, specificity, affinity; recommended ranges: coating antibody 1-15 µg/mL, detection antibody 0.5-10 µg/mL depending on purity [30] [31] |
| Solid Phase | Polystyrene microplates (high, medium, low protein binding) | Immobilization of capture antibody or antigen | Binding capacity, surface characteristics, well geometry |
| Buffers | Coating buffer (e.g., carbonate, PBS), Blocking buffer (e.g., BSA, casein, non-fat milk), Wash buffer (e.g., PBS-Tween) | pH maintenance, reduction of non-specific binding, removal of unbound materials | Composition, pH, ionic strength, detergent concentration |
| Detection System | HRP or AP enzyme conjugates, Chromogenic/chemiluminescent substrates (e.g., TMB, ABTS), Stop solution (e.g., sulfuric acid) | Signal generation and amplification | Enzyme concentration (HRP: 0.02-0.2 µg/mL; AP: 0.1-0.2 µg/mL), substrate sensitivity, linear range of detection [30] [31] |
| Reference Materials | Certified reference standards, Control samples (positive, negative) | Calibration, quality control, validation | Matrix matching, stability, concentration assignment |
Data Analysis and Quality Control
Quantitative Analysis of Optimization Results
Both checkerboard titration and Taguchi Method optimization require systematic quantitative analysis to identify optimal conditions. For checkerboard titration, data is typically analyzed by calculating signal-to-noise ratios for each well and identifying the combination that maximizes this ratio while maintaining an appropriate dynamic range [8]. For the Taguchi Method, the primary analysis involves calculating the average signal-to-noise ratio for each factor at each tested level, then selecting the level that produces the highest average S/N ratio for each individual factor [32]. The optimal condition is the combination of these individual factor levels. Statistical software packages can facilitate this analysis, particularly for Taguchi experiments with multiple factors and complex interactions.
Table 2: Key Validation Parameters for Optimized ELISA Protocols
| Validation Parameter | Target Value | Experimental Approach | Interpretation |
|---|---|---|---|
| Accuracy (Recovery) | 80-120% recovery | Spike-and-recovery experiments: add known analyte amounts to sample matrix [8] [31] | Measures matrix effects; values close to 100% indicate minimal interference |
| Precision | Intra-assay CV <10%, Inter-assay CV <15% | Repeated measurements of same sample within and between runs [31] | Lower CV% indicates higher reproducibility |
| Sensitivity (Limit of Detection) | Typically 2-3 SD above background | Analysis of zero standard replicates; lowest concentration distinguishable from background | Defines the lowest detectable analyte level |
| Dynamic Range | 2-3 logs of concentration | Serial dilution of standard; linear range of standard curve | Working range of the assay where quantification is accurate |
| Specificity | Minimal cross-reactivity (<5%) | Testing against structurally similar molecules | Ability to distinguish target from related molecules |
| Dilution Linearity | %CV <15-20% across dilutions | Serial dilution of high-concentration sample [8] [31] | Confirms accurate measurement at different sample concentrations |
Troubleshooting and Quality Assessment
Even after systematic optimization, ELISA protocols may require additional refinement. Common issues include high background signals (often addressed by optimizing blocking conditions or antibody concentrations), poor sensitivity (improved through incubation time optimization or enhanced detection systems), and high variability between replicates (typically resolved by standardizing washing procedures and reagent handling) [29] [33]. After identifying optimal conditions through checkerboard titration or Taguchi Methods, comprehensive validation should include assessment of the standard curve performance with an R² value >0.98 for 4-parameter logistic (4PL) curve fitting, which is widely recommended for ELISA data analysis [33]. Additional quality control measures include testing parallelism between the standard curve and diluted samples to detect matrix effects, and establishing appropriate acceptance criteria for control samples in each assay run [8]. These systematic approaches to optimization and validation collectively ensure development of robust, reliable ELISA methods suitable for research and diagnostic applications.
Application Note: In-house SARS-CoV-2 IgG Serology Assay
Serology tests are valuable tools for surveillance of virus circulation and evaluation of protective immunity prevalence among populations, which is crucial for public health strategies and "exit strategies" from pandemic restrictions [34]. This application note details the development and optimization of an in-house enzyme-linked immunosorbent assay (ELISA) for detecting SARS-CoV-2 specific IgG antibodies, evaluated against the micro-neutralization (MN) assay as a reference test [34].
Materials and Methods
Key Research Reagent Solutions are listed in Table 1.
Table 1: Essential Reagents for SARS-CoV-2 IgG ELISA
| Reagent / Material | Function in the Assay |
|---|---|
| SARS-CoV-2 full-length Spike (S) ECD-His Recombinant Protein | Antigen used to coat plates for capturing virus-specific antibodies from serum samples [34]. |
| High-Binding ELISA Plates | Solid surface for immobilizing the capture antigen [34]. |
| Human Serum Samples | Test specimens for the presence of anti-SARS-CoV-2 IgG antibodies [34]. |
| Anti-Human IgG Antibody conjugated with Horseradish Peroxidase (HRP) | Detection antibody that binds to captured human IgG; enzyme conjugate for signal generation [34]. |
| Tetramethylbenzidine (TMB) Substrate | Chromogenic substrate for HRP; produces a measurable color change upon enzymatic reaction [34]. |
| Micro-neutralization (MN) Assay | Gold standard reference test used to determine the true sero-status of samples and validate ELISA performance [34]. |
Optimized Protocol:
- Coating: ELISA plates were coated with 100 ng per well of SARS-CoV-2 full-length spike (S) recombinant protein [34].
- Blocking: Plates were blocked with an appropriate protein-based buffer (e.g., BSA or serum) to prevent non-specific binding [35].
- Sample Incubation: Human serum samples, diluted at 1:100, were added to the plates and incubated [34].
- Detection: A horseradish peroxidase (HRP)-conjugated anti-human IgG antibody, diluted at 1:64,000, was used for detection [34].
- Signal Measurement: The reaction with TMB substrate was stopped, and the optical density (OD) was measured at 450 nm [34].
Performance Evaluation: The assay was evaluated using 418 human serum samples (109 positive, 309 negative) as determined by the MN assay (MN titer ≥ 1:20 considered positive) [34].
Results and Data Analysis
The performance characteristics of the optimized in-house SARS-CoV-2 IgG ELISA are summarized in Table 2.
Table 2: Performance Metrics of the SARS-CoV-2 IgG ELISA
| Performance Metric | Result |
|---|---|
| Sensitivity | 100% |
| Specificity | 98.4% |
| Overall Agreement | 98.8% |
| Preliminary Cut-off Value (OD₄₅₀) | 0.27 |
| ROC Curve Analysis (Optimal Cut-off) | 0.29 (providing 100% sensitivity and 98.54% specificity) |
| Area Under the Curve (AUC) | 0.9996 |
A statistically significant positive correlation was observed between the ELISA OD450 values and the MN titers of positive samples (rs = 1, p-value = 0.016), indicating that the assay signal correlates with neutralizing antibody levels [34]. The assay demonstrated high specificity for SARS-CoV-2, as sera containing antibodies against MERS-CoV or HCoV-HKU1 tested negative [34].
Application Note: Cytokine Quantification via Sandwich ELISA
Cytokines are crucial signaling molecules in inflammation, and their precise quantification is essential for understanding disease processes, immune responses, and drug mechanisms [35]. The sandwich ELISA is a widely used method for detecting and quantifying specific cytokines in complex biological samples due to its high specificity and sensitivity, often in the picogram per milliliter range [35] [36].
Materials and Methods
Key Research Reagent Solutions are listed in Table 3.
Table 3: Essential Reagents for Cytokine Sandwich ELISA
| Reagent / Material | Function in the Assay |
|---|---|
| Matched Antibody Pair (Capture & Detection) | Antibody pair that binds to distinct epitopes on the target cytokine, providing high specificity [35]. |
| Recombinant Cytokine Protein | Used to generate the standard curve for quantitative analysis [35]. |
| Biotinylated Detection Antibody | Detection antibody labeled with biotin for subsequent amplification [35]. |
| Streptavidin-Horseradish Peroxidase (SA-HRP) | Enzyme conjugate that binds to biotin; amplifies the detection signal [36]. |
| Enhanced Protein-Binding ELISA Plates | Plates with high binding capacity for optimal adsorption of the capture antibody [36]. |
| Blocking Buffer (e.g., BSA or Serum) | Prevents non-specific binding of proteins to any remaining plastic surface [36]. |
Detailed Protocol: The following protocol, adapted from a standard cytokine ELISA procedure, typically requires two days to complete [36].
Day 1:
- Coating: Dilute the purified capture antibody to 1-4 µg/mL in a binding solution (e.g., 0.1 M Sodium Phosphate, pH 9.0). Add 100 µL per well to a 96-well plate and incubate overnight at 4°C [36].
- Blocking: Remove the coating solution and add 200 µL of blocking buffer (e.g., 1% BSA or 10% serum in PBS) per well. Incubate at room temperature for 1-2 hours [36].
- Washing: Wash the plate ≥3 times with wash buffer (e.g., PBS with 0.05% Tween-20) [36].
Day 2:
- Standards and Samples: Prepare serial dilutions of the recombinant cytokine standard and dilute samples in an appropriate buffer. Add 100 µL per well in duplicate. Incubate for 2-4 hours at room temperature or overnight at 4°C [36].
- Washing: Wash the plate ≥4 times [36].
- Detection Antibody: Add 100 µL per well of the biotinylated detection antibody (diluted to 0.5-2 µg/mL). Incubate for 1 hour at room temperature [36].
- Washing: Wash the plate ≥4 times [36].
- Enzyme Conjugate: Add 100 µL per well of pre-titered Streptavidin-HRP. Incubate for 30 minutes at room temperature [36].
- Washing: Wash the plate ≥5 times [36].
- Substrate Development: Add 100 µL per well of TMB substrate. Incubate in the dark for 5-30 minutes for color development [36].
- Stop and Read: Stop the reaction with an equal volume of stop solution (e.g., 1.5 N Sulfuric Acid) and read the optical density immediately at 405 nm (for TMB after acid stop) or 450 nm (for TMB without stopping) [36].
Results and Data Analysis
Standard Curve and Quantification: A standard curve is generated by plotting the mean absorbance (y-axis) against the cytokine concentration (x-axis) of the serial dilutions [18]. The concentration of cytokine in unknown samples is interpolated from this curve. Data reduction methods such as 4-parameter logistic (4PL) or 5-parameter logistic (5PL) curve fitting are recommended for optimal results as they account for the non-linear, sigmoidal nature of ELISA data [9]. An example of a standard curve is shown in Figure 1.
Assay Validation:
- Precision: Samples should be run in duplicate or triplicate. The coefficient of variation (CV) between replicates should be ≤20% to ensure reproducibility [18] [9].
- Spike Recovery: A known concentration of the cytokine is spiked into the sample matrix and quantified. Recovery close to 100% indicates minimal matrix interference [9].
Troubleshooting Common ELISA Problems and Proven Optimization Techniques
Enzyme-linked immunosorbent assay (ELISA) remains a cornerstone technique for protein biomarker detection in research and diagnostic laboratories. However, signal-related issues—whether weak signal, excessive signal, or high background—represent significant challenges that compromise data reliability and experimental outcomes. These problems often stem from subtle deviations in protocol execution, reagent quality, or assay optimization. Within the broader context of ELISA protocol optimization research, systematic troubleshooting is essential for achieving robust, reproducible results. This application note provides a comprehensive framework for diagnosing and resolving the most prevalent ELISA signal abnormalities, enabling researchers to enhance assay sensitivity, specificity, and overall performance.
Table of Common ELISA Signal Issues and Solutions
| Problem Category | Specific Symptom | Possible Causes | Recommended Solutions |
|---|---|---|---|
| Weak or No Signal | Low absorbance readings, flat standard curve | Reagents not at room temperature [37], incorrect antibody dilutions [37], expired reagents [37], insufficient incubation times [38], plate read at incorrect wavelength [37] | Allow all reagents to warm for 15-20 minutes before use [37], verify antibody dilution calculations [37], check reagent expiration dates [37], ensure adequate incubation times [38], confirm correct wavelength on plate reader [37] |
| Excessive Signal | Saturation signal at high OD, poor standard curve linearity | Excessive antibody concentrations [39], prolonged substrate incubation [37] [39], substrate contamination or degradation [40] | Titrate antibodies to optimal concentration [39], strictly follow recommended substrate incubation times [37], prepare fresh substrate and protect from light [40] |
| High Background | Elevated signal across all wells including blanks | Inadequate washing [37] [40], insufficient blocking [41] [42], non-specific antibody binding [40] [39], cross-reactivity [40] [42], contaminated reagents [40] [42] | Increase wash cycles and incorporate soak steps [37], optimize blocking buffer concentration and duration [41] [42], use pre-adsorbed secondary antibodies [39], ensure high antibody specificity [40], use fresh, high-quality water and buffers [40] [42] |
| Poor Replicate Data | High CV values, inconsistent duplicates | Inconsistent pipetting technique [9], plate sealing issues [37], temperature fluctuations during incubation [37] | Calibrate pipettes and ensure proper tip sealing [9], use fresh plate sealers for each incubation [37], maintain stable incubation temperature away from drafts [37] [9] |
Experimental Protocols for Signal Issue Diagnosis
Protocol 1: Systematic Approach to High Background Troubleshooting
High background remains one of the most persistent challenges in ELISA development, often resulting from multiple contributing factors. This protocol provides a step-by-step diagnostic approach.
Materials:
- Coated ELISA plate
- Appropriate blocking buffers (e.g., BSA, casein, commercial StabilGuard formulations [40])
- Wash buffer (PBS or Tris-based with Tween-20)
- Primary and secondary antibodies
- Substrate and stop solutions
- Plate reader
Method:
- Evaluate Washing Efficiency:
Assess Blocking Efficiency:
Investigate Antibody Specificity:
Check Substrate System:
Interpretation: Significant reduction in background after implementing any step indicates the identified area as the primary contributor to high background. For persistent issues, consider sample-specific effects or matrix interference.
Protocol 2: Optimization for Weak Signal Enhancement
Weak signals compromise assay sensitivity and limit detection of low-abundance targets. This protocol addresses common causes of insufficient signal.
Materials:
- Antigen standard
- Coated ELISA plate
- Detection antibodies
- Signal amplification reagents (if applicable)
- High-sensitivity substrate
Method:
- Verify Reagent Preparation:
- Confirm all reagents have reached room temperature (15-20 minutes) before starting assay [37].
- Double-check dilution calculations for standards and antibodies.
- Ensure proper reconstitution of standard materials.
Optimize Incubation Conditions:
Evaluate Signal Generation System:
Assess Plate Coating Efficiency:
- For in-house coated plates, confirm use of ELISA-specific plates (not tissue culture plates) [37].
- Verify coating buffer is carbonate/bicarbonate (pH 9.6) for optimal antibody adsorption.
- Test different coating antibody concentrations if developing in-house assay.
Interpretation: Systematic implementation of these steps typically identifies the limiting factor in signal generation. Signal amplification methods generally provide the most significant enhancement for low-abundance targets.
Advanced Optimization Strategies
Surface Engineering for Enhanced Performance
Traditional passive adsorption of capture antibodies can lead to random orientation and partial denaturation, reducing assay sensitivity. Advanced surface modification strategies address these limitations:
Oriented Immobilization:
- Protein A/G Coating: Utilize the Fc-binding properties of Protein A or G to ensure proper antibody orientation, increasing antigen-binding capacity [41].
- Biotin-Streptavidin System: Biotinylate capture antibodies for precise orientation via streptavidin-coated plates [41].
- Covalent Immobilization: Employ crosslinkers for stable antibody attachment, preventing antibody loss during washes [41].
Non-fouling Surface Modifications:
- Implement polyethylene glycol (PEG) polymer coatings or polysaccharides (chitosan, dextran) to minimize non-specific binding [41].
- These modifications create a bioinert surface that resists protein adsorption, significantly improving signal-to-noise ratios [41].
Innovative Signal Amplification Approaches
Bridging the sensitivity gap between ELISA and nucleic acid tests requires advanced signal generation strategies:
Cell-Free Synthetic Biology:
- Expression Immunoassays: Integrate in vitro transcription and translation systems to generate reporter proteins in response to target detection [41].
- CLISA (CRISPR-Linked Immunoassays): Employ CRISPR systems to amplify signal through nucleic acid detection cascades [41].
- TLISA (T7 RNA Polymerase–Linked Immunosensing Assays): Utilize T7 RNA polymerase amplification for ultra-sensitive detection [41].
Nanomaterial-Enhanced Detection:
- Incorporate gold nanoparticles, quantum dots, or enzymatic nanomaterials to amplify signals while maintaining low background [43].
- These approaches can achieve detection limits in the attomolar range, significantly surpassing conventional ELISA [43].
Visualization of ELISA Troubleshooting Workflow
ELISA Troubleshooting Decision Pathway
This workflow provides a systematic approach to diagnosing and resolving common ELISA signal issues. The logical progression from problem identification through verification ensures comprehensive troubleshooting while minimizing unnecessary procedural changes.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent Category | Specific Products | Function in ELISA Optimization |
|---|---|---|
| Blocking Buffers | StabilGuard [40], Bovine Serum Albumin (BSA) [41], Casein [41], Normal Serum [39] | Reduce non-specific binding by occupying uncovered surface areas on the plate, critical for lowering background [41] [40]. |
| Sample/Assay Diluents | MatrixGuard [40], Surmodics Assay Diluent [40] | Block matrix interferences in complex samples (e.g., serum, plasma) while maintaining true assay signal [40]. |
| Wash Buffers | PBS with 0.05% Tween-20 [42], Tris-buffered Saline with Tween-20 | Remove unbound reagents during washing steps; detergent concentration critical for minimizing background [42]. |
| Detection Substrates | BioFX TMB [40], Chemiluminescent substrates [40], pNPP (for alkaline phosphatase) [38] | Generate measurable signal through enzyme-catalyzed reaction; selection impacts sensitivity, dynamic range, and background [38] [40]. |
| Plate Sealers | Adhesive plate seals [37] | Prevent well-to-well contamination and evaporation during incubations; fresh sealers recommended for each step [37]. |
| Orientation Reagents | Protein A/G [41], Biotin-Streptavidin systems [41] | Improve antibody binding capacity through Fc-specific orientation, enhancing assay sensitivity [41]. |
Effective troubleshooting of ELISA signal issues requires a systematic approach that addresses both technical execution and fundamental assay design. By implementing the diagnostic protocols and optimization strategies outlined in this application note, researchers can significantly enhance assay performance, particularly for challenging applications requiring high sensitivity and precision. The integration of advanced surface chemistry approaches and innovative signal amplification technologies represents the future of ELISA development, potentially bridging the sensitivity gap with molecular detection methods. As ELISA continues to evolve within diagnostic and drug development pipelines, rigorous optimization and troubleshooting methodologies will remain essential for generating reliable, reproducible data in protein biomarker detection and quantification.
The Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technology for detecting and quantifying specific proteins in research and drug development. However, the reliability of experimental conclusions hinges entirely on the quality of the underlying ELISA data. Poor replicate data, suboptimal standard curves, and inconsistent results between assays represent three of the most pervasive challenges threatening data integrity. These issues can obscure true biological signals, lead to erroneous interpretations, and compromise the reproducibility of scientific findings. This application note systematically addresses the root causes of these data quality problems and provides detailed, actionable protocols for their resolution, framed within the critical context of ELISA protocol optimization research. A proactive approach to assay validation is not merely a best practice but a fundamental requirement for generating robust, publication-quality data.
Troubleshooting Common Data Quality Issues
A systematic approach to troubleshooting is essential for diagnosing and resolving common ELISA data problems. The table below outlines frequent issues, their potential causes, and recommended solutions.
Table 1: Troubleshooting Common ELISA Data Quality Problems
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| Poor Replicate Data (High CV >20%) [44] | Pipetting errors; Insufficient washing [37]; Contamination; Evaporation [44] | Check pipette calibration/technique; Optimize wash procedure (volume, soak time, cycles) [37]; Use fresh plate sealers [37]; Ensure stable incubation temperature [44]. |
| Poor Standard Curve | Incorrect serial dilution; Capture antibody not properly bound [37]; Unstable incubation temperature [37] | Verify pipetting technique and calculations [37]; Ensure use of correct plate type and coating buffer [37]; Use a 4-parameter logistic (4PL) algorithm for curve fitting [44]. |
| High Background | Insufficient washing [37]; Substrate exposed to light [37]; Non-specific antibody binding | Increase wash cycles and duration; Add soak steps [37]; Store substrate in dark; Optimize blocking buffer concentration and incubation [7] [8]. |
| Assay-to-Assay Variability | Inconsistent reagent preparation; Temperature fluctuations [37]; Operator technique; Different reagent lots | Aliquot and standardize reagents; Use controls on every plate [44]; Adhere strictly to incubation times; Validate new reagent lots [8]. |
| Weak or No Signal | Reagents not at room temperature; Expired reagents; Incorrect detector antibody dilution [37] | Equilibrate all reagents to room temperature before assay [37]; Confirm reagent expiration dates [37]; Titrate detection antibody for optimal concentration [7]. |
The Critical Role of Replicates and Controls
Understanding the purpose and execution of replicates and controls is the first defense against poor data quality.
Types of Replicates:
- Technical Replicates: Multiple measurements of the same biological sample. They assess the variability of the assay procedure itself (pipetting, washing, etc.) [45].
- Biological Replicates: Measurements from different biological specimens. They account for natural biological variation and form the bedrock of sound statistical analysis [45].
Choosing Replicate Number:
- Singles: Suitable only for high-throughput qualitative screening or when throughput is paramount and statistical power comes from a large number of unique biological samples. They do not allow for error detection and are not recommended for quantitative analysis [45].
- Duplicates: The ideal compromise for most quantitative ELISAs. They enable error detection by calculating the % Coefficient of Variation (%CV), with a common acceptability threshold of <15-20% [44] [18]. If the CV exceeds this threshold, the sample should be retested. However, duplicates do not allow for the identification of which specific well is an outlier [45].
- Triplicates: Provide the highest level of precision and allow for outlier identification and exclusion based on predefined statistical criteria. This comes at the cost of significantly reduced throughput and higher reagent consumption. Their use is indicated when data precision is paramount [45].
Essential Controls:
- Blank: Contains coating and blocking buffer only; measures signal from the plate and blocking buffer [46].
- Zero Standard (ZC): Contains all assay reagents and the sample matrix without the target analyte; defines the true "background" of the assay [46].
- Positive Control: A known concentration of the target analyte; verifies the assay is performing as expected [8].
- Standard Curve: A dilution series of known analyte concentrations, run on every plate, is non-negotiable for accurate quantification [44].
Experimental Protocols for Optimization and Validation
The following protocols provide detailed methodologies for key optimization and validation experiments essential for resolving data quality issues.
Checkerboard Titration for Antibody Optimization
This protocol is designed to simultaneously optimize the concentrations of the capture and detection antibodies, which is fundamental to achieving a strong signal-to-noise ratio [7] [8].
Methodology:
- Plate Coating: Prepare a dilution series of the capture antibody in coating buffer (e.g., PBS). Refer to Table 2 for recommended starting concentrations. Dispense different concentrations of capture antibody across the rows of a 96-well ELISA plate (e.g., 100 µL/well).
- Incubation and Blocking: Cover the plate and incubate overnight at 4°C or for 1-2 hours at room temperature. Wash the plate 3 times with wash buffer. Block the plate with an appropriate blocking buffer (e.g., 1% BSA in PBS) for 1-2 hours at room temperature.
- Antigen Addition: Wash the plate 3 times. Add a fixed, moderate concentration of the target antigen (or a positive control sample) to all wells. Incubate for 2 hours at room temperature.
- Detection Antibody Titration: Wash the plate 3 times. Prepare a dilution series of the detection antibody in blocking buffer. Dispense different concentrations of the detection antibody down the columns of the plate.
- Signal Development: After incubation and washing, add the enzyme-conjugated secondary antibody (if needed), incubate, wash, and add the substrate. Stop the reaction and read the absorbance.
- Analysis: Identify the combination of capture and detection antibody concentrations that yields the strongest signal for the positive control with the lowest background (signal from zero standard).
Table 2: Recommended Antibody Concentration Ranges for Checkerboard Titration [7]
| Antibody Source | Coating Antibody Range (µg/mL) | Detection Antibody Range (µg/mL) |
|---|---|---|
| Polyclonal Serum | 5 – 15 | 1 – 10 |
| Crude Ascites | 5 – 15 | 1 – 10 |
| Affinity-Purified Polyclonal | 1 – 12 | 0.5 – 5 |
| Affinity-Purified Monoclonal | 1 – 12 | 0.5 – 5 |
Protocol for Spike-and-Recovery and Linearity of Dilution
This validation protocol assesses matrix interference, which is a common cause of poor standard curves and inaccurate sample quantification [8].
Spike-and-Recovery Methodology:
- Sample Preparation:
- Background Sample: Use the actual sample matrix (e.g., serum, cell culture media) known to be free of the target analyte, or a suitable surrogate.
- Spiked Sample: Spike a known, moderate concentration of the purified standard analyte into the background sample.
- Reference Sample: Spike the same concentration of standard analyte into the diluent used to prepare the standard curve.
- Assay Execution: Run the spiked sample, the reference sample, and a standard curve in the same ELISA.
- Calculation and Analysis:
- Calculate the measured concentration of the spiked sample and the reference sample from the standard curve.
- % Recovery = (Concentration of Spiked Sample / Concentration of Reference Sample) x 100.
- A recovery of 80-120% is generally acceptable, indicating minimal matrix interference [8].
Dilutional Linearity Methodology:
- Sample Preparation: Select a sample with a high endogenous concentration of the analyte. Perform a series of serial dilutions (e.g., 1:2, 1:4, 1:8, etc.) using the recommended assay diluent.
- Assay Execution: Measure the concentration of each diluted sample using the ELISA standard curve.
- Analysis: Plot the observed concentration against the expected concentration (based on the dilution factor). The plot should be linear. A loss of linearity (e.g., curves downward) indicates matrix effects, suggesting the need for optimization of the sample diluent [8].
Data Analysis Best Practices
Proper data analysis is the final, critical step in ensuring data quality. Adherence to the following practices mitigates the risk of introducing errors during the analysis phase.
- Standard Curve on Every Plate: Environmental conditions, incubation times, and operator technique can vary daily. Running a fresh standard curve on each plate is essential for accurate quantification and is a non-negotiable best practice [44].
- Correct Curve-Fitting Algorithm: The complex binding kinetics of immunoassays are best modeled by a 4-parameter logistic (4PL) regression curve. This algorithm accounts for the asymmetric sigmoidal shape of the standard curve and provides the most accurate fit, especially at the upper and lower asymptotes [44] [18]. Avoid using simple linear regression.
- Background Subtraction: Subtract the average optical density (OD) of the zero standard (or blank) wells from all other standard and sample readings. This corrects for background signal inherent to the assay system [44] [18].
- Replicate Analysis: Calculate the mean, standard deviation (SD), and %CV for all replicates. A %CV ≤ 20% is a widely accepted benchmark for pipetting precision and assay robustness. Investigate any samples with a higher %CV [44] [18].
- Account for Dilution Factor: When reporting final concentrations, remember to multiply the concentration derived from the standard curve by the sample's dilution factor [44] [18].
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential materials and reagents critical for successful ELISA development and troubleshooting.
Table 3: Key Research Reagent Solutions for ELISA Optimization
| Reagent / Material | Function & Importance in Optimization | Key Considerations |
|---|---|---|
| Matched Antibody Pairs | Capture and detection antibodies that bind to non-overlapping epitopes of the target antigen. Fundamental for sandwich ELISA development [7]. | Specificity and affinity are critical. Must be validated for pair compatibility to ensure efficient "sandwiching." |
| ELISA Microplates | Solid surface for immobilization of the capture antibody [37]. | Use high-binding plates (e.g., Nunc MaxiSorp) [47]. Tissue culture-treated plates are not suitable [37]. |
| Blocking Buffer | Prevents non-specific binding of proteins to any remaining plastic surface, reducing background [7] [8]. | BSA or casein-based solutions are common. Concentration (1-5%) and protein source may require optimization [7]. |
| Coating Buffer | Solution (typically carbonate/bicarbonate or PBS) used to dilute the capture antibody for plate immobilization [8]. | pH and ionic strength can affect antibody binding. Ensure correct preparation. |
| Wash Buffer | Removes unbound reagents, reducing background and non-specific signal [37] [46]. | Typically PBS or Tris with a mild detergent (e.g., 0.05% Tween-20). Soak time and cycle number are key optimization parameters [37]. |
| Enzyme Conjugate | Enzyme-linked antibody (e.g., HRP or AP) that catalyzes substrate conversion for detection [7]. | Concentration must be optimized (see Table 4). Too high causes background, too low weakens signal [7]. |
| Detection Substrate | Chromogenic, chemiluminescent, or fluorescent molecule converted by the enzyme to a detectable signal [7] [3]. | Choice depends on required sensitivity and available instrumentation. Protect from light [37]. |
Table 4: Recommended Enzyme Conjugate Concentrations for Different Detection Systems [7]
| Enzyme | Detection System | Typical Concentration Range (ng/mL) |
|---|---|---|
| HRP | Colorimetric | 20 – 200 |
| HRP | Chemifluorescent | 25 – 50 |
| HRP | Chemiluminescent | 10 – 100 |
| AP | Colorimetric | 100 – 200 |
| AP | Chemiluminescent | 40 – 200 |
In enzyme-linked immunosorbent assay (ELISA) protocol optimization, the washing steps are frequently undervalued despite being critical determinants of assay performance. Effective washing minimizes non-specific background signal, thereby enhancing the specificity, sensitivity, and overall reliability of the assay [48]. Incomplete removal of unbound antibodies, antigens, or enzyme conjugates during washing procedures is a primary contributor to high background noise, which can obscure true positive signals and compromise data interpretation [49] [50]. This application note details optimized washing protocols and techniques designed to maximize signal-to-noise ratio, providing researchers and drug development professionals with methodologies to significantly improve ELISA outcomes.
The Consequences of Inadequate Washing
Insufficient washing directly undermines assay specificity by failing to remove non-specifically bound detection antibodies. These retained antibodies generate false-positive signals that reduce the signal-to-noise ratio and can lead to inaccurate quantification of the target analyte [50]. Non-specific binding is particularly problematic in complex biological matrices such as serum or blood, where interferent proteins can adhere to the assay substrate or plate surface [41] [50].
Advanced single-molecule studies have quantified the dramatic impact of non-specific binding. In model systems, high concentrations of detection antibody (500 nM) in the absence of target antigen produced approximately 92 non-specifically bound detection antibody molecules per field of view. Implementing stringent washing and validation procedures is essential to mitigate this background [50].
Optimization of Washing Buffers
The composition of the washing buffer is fundamental to achieving effective removal of unbound reagents while maintaining the stability of specifically formed antigen-antibody complexes.
Standard Buffer Formulations
Table 1: Common Washing Buffer Compositions for ELISA
| Component | PBS-Based Buffer | TBS-Based Buffer | Function |
|---|---|---|---|
| Buffer Base | 0.01M Phosphate Buffered Saline (PBS), pH 7.4 | 0.05M Tris-Buffered Saline (TBS), pH 8.0 | Maintains physiological pH and ionic strength |
| Detergent | 0.05-0.1% Tween 20 | 0.05-0.1% Tween 20 | Reduces surface tension, disrupts hydrophobic interactions |
| Alternative Detergent | 0.1% Triton X-100 (PBST) | 0.1% Triton X-100 (TBST) | Alternative non-ionic detergent for challenging applications |
The optimal pH and ionic strength of the buffer base stabilizes antibody-antigen complexes while facilitating the removal of unbound reagents [48]. The inclusion of a non-ionic detergent, most commonly Tween 20 at concentrations between 0.05% and 0.1%, is critical for reducing surface tension and disrupting hydrophobic interactions that cause non-specific binding [48]. In some cases, alternative detergents like Triton X-100 may be used to improve solubility or specificity for particular antigens or antibodies [48].
Washing Procedure and Technique
The mechanical process of washing is as important as buffer composition. Consistency and thoroughness in technique are vital for well-to-well reproducibility [51].
Manual Washing Protocol
- Aspiration: Completely aspirate liquid from all wells by gently lowering an aspiration tip to the bottom of the well. Avoid scratching the well surface, as physical damage can increase non-specific binding [49].
- Dispensing: Fill each well completely with washing buffer. A volume of 300-400 µL per well for a 96-well plate is typically sufficient to displace any residual liquid.
- Soaking: Allow the buffer to stand in the wells for 10-30 seconds to dissociate loosely bound materials.
- Repetition: Repeat the cycle. Most protocols require 3-5 wash cycles per washing step [48]. For persistent background, increasing the number of washes or the duration of soaks (e.g., 10-minute washes) can be highly effective [48].
- Final Removal: After the final wash, invert the plate and tap it firmly onto absorbent laboratory tissue to remove any residual fluid [49].
Automated Washing Systems
Automated plate washers provide superior consistency and efficiency by standardizing the aspiration and dispensing pressure, volume, and pattern across all wells [3]. This minimizes user-to-user variability and is particularly advantageous for high-throughput applications. The program should be configured to ensure complete well coverage and efficient fluid removal.
Integrated Workflow and Strategic Washing Placement
Washing is not an isolated activity but an integral component of the entire ELISA workflow. Its effectiveness is influenced by prior steps, particularly blocking.
The Researcher's Toolkit: Essential Reagents for Optimized Washing
Table 2: Key Research Reagent Solutions for ELISA Washing Optimization
| Reagent / Material | Function & Purpose | Optimization Notes |
|---|---|---|
| Washing Buffer (e.g., PBST) | Removes unbound reagents; critical for low background. | Detergent concentration (0.05-0.1% Tween 20) is key [48]. |
| Automated Plate Washer | Provides consistent, reproducible washing across all wells. | Reduces user variability; essential for high-throughput screens [3]. |
| Blocking Buffer (e.g., BSA, Casein) | Pre-analytic step that coats non-specific binding sites on the plate. | Ineffective blocking compromises washing efficiency; choose blocker compatible with sample and antibodies [41] [48]. |
| High-Affinity, Specific Antibodies | Primary reagents for target capture and detection. | Affinity-purified antibodies significantly reduce non-specific binding, making unwanted interactions easier to wash away [49] [51]. |
Troubleshooting High Background
A structured approach is essential for diagnosing and resolving washing-related issues.
If high background persists despite optimized washing, investigate these potential culprits:
- Detection Antibody Concentration: Excessive detection antibody is a major source of background. Titrate to find the concentration that gives the strongest specific signal with the lowest background [49] [50].
- Enzyme Conjugate Concentration: Over-concentration of enzyme conjugate (e.g., HRP-Streptavidin) can saturate the system. Use the recommended concentration range (20-200 ng/mL for HRP in colorimetric systems) and titrate if necessary [7].
- Blocking Efficiency: Inadequate blocking leaves reactive sites on the plate. Ensure blocking is complete by testing different blocking agents (BSA, casein, milk) and concentrations (1-5%) [41] [48].
Rigorous optimization of washing techniques is a critical and non-negotiable aspect of robust ELISA development. By implementing structured protocols for buffer composition, mechanical process, and integrated workflow strategy, researchers can achieve the high specificity and low background essential for reliable, publication-quality data.
Within the broader context of Enzyme-Linked Immunosorbent Assay (ELISA) protocol optimization, controlling for environmental and procedural variables is a fundamental prerequisite for assay robustness and data integrity. These factors, if unmanaged, introduce significant variability that can compromise the accuracy and reproducibility of results, even in otherwise meticulously designed experiments. This application note details standardized protocols for identifying, quantifying, and mitigating the effects of three critical challenges: temperature fluctuations, solvent evaporation, and edge effects. The procedures outlined herein are designed to provide researchers and drug development professionals with actionable strategies to enhance the reliability of their immunoassays.
Core Principles and Impact on Assay Performance
Environmental and procedural factors interfere with assay performance by disrupting the biochemical kinetics of the antigen-antibody interaction, modifying the physical state of reagents, and introducing spatial bias across the microplate. Table 1 summarizes the primary causes and consequences of these key variables.
Table 1: Impact of Environmental and Procedural Variables on ELISA Performance
| Variable | Primary Causes | Consequences on Assay Performance |
|---|---|---|
| Temperature | Incubation outside recommended range, fluctuating incubator temperatures, improper reagent pre-warming [52] [53]. | Altered antibody-antigen binding kinetics; increased non-specific binding; compromised enzyme activity and stability; significant shifts in standard curves and sample quantitation [52] [53]. |
| Evaporation | Inadequate plate sealing, prolonged incubations, low ambient humidity [54]. | Increased well-to-well concentration variability; elevated background signal; precipitation of salts and proteins [54]. |
| Edge Effects | Temperature gradients across the microplate during incubation, uneven washing [52] [54]. | Higher intra-assay coefficient of variation (%CV); inconsistent results between edge and interior wells; invalidated standard curve [52] [54]. |
The following diagram illustrates the interconnected nature of these variables and their collective impact on the final assay readout.
Experimental Protocols for Control and Validation
Protocol 1: Temperature Kinetics and Stability Profiling
This protocol systematically evaluates the impact of temperature on key assay reagents and the overall immunoassay reaction.
- Objective: To determine the optimal and stable incubation temperature for the assay and to establish the thermal stability of critical reagents.
- Materials:
- Validated ELISA kit (e.g., Leptin Ultrasensitive, ALPCO) or in-house assay components [53].
- Precision incubators or thermal cyclers set to 4°C, 22°C (room temperature), 37°C, and 40°C.
- Pre-characterized positive control sample (e.g., pooled serum, recombinant protein).
- Microplate reader.
- Method:
- Reagent Stability Test: Divide a single lot of capture antibody, detection antibody, and enzyme conjugate into aliquots. Incubate separate aliquots at each test temperature (4°C, 22°C, 37°C, 40°C) for 24 hours. After treatment, return all aliquots to 4°C.
- Assay Performance Test: Coat plates with a standardized concentration of capture antibody. Perform a complete ELISA using the temperature-treated reagents and the positive control sample. Run each temperature condition on a separate plate, with all plates containing a full standard curve and the positive control in replicates of n=6.
- Incubation Optimization: Using untreated reagents, run the assay but vary the incubation temperature for the key antigen-antibody binding step (e.g., sample incubation) across the test temperatures. Keep all other incubation steps constant.
- Data Analysis: For each condition, calculate the mean absorbance for the positive control, the background signal, the signal-to-noise ratio, and the %CV of the replicates. Plot the standard curve for each condition and compare the calculated concentration of the positive control.
- Validation Criterion: The optimal temperature condition is that which yields the highest signal-to-noise ratio, a %CV of <10% for replicate wells, and a standard curve with an R² value >0.99.
Protocol 2: Evaporation and Edge Effect Mapping
This protocol quantifies the spatial bias introduced by evaporation and temperature gradients across a microplate.
- Objective: To visualize and quantify the "edge effect" and to validate sealing methods for preventing evaporation.
- Materials:
- Clear, flat-bottom 96-well microplate.
- Two different plate sealers (e.g., adhesive film vs. silicone cap mat).
- A solution of a stable colorimetric dye (e.g., TMB stop solution).
- Microplate reader.
- Method:
- Plate Setup: Fill all 96 wells with an identical volume (e.g., 100 µL) of the dye solution.
- Sealing and Incubation: Seal one plate with Adhesive Sealer A and a second plate with Sealer B. Leave a third plate unsealed. Incubate all plates for the duration of a typical ELISA protocol (e.g., 2-4 hours) in a 37°C incubator.
- Data Acquisition: Read the absorbance of all wells at the appropriate wavelength for the dye.
- Data Analysis: Calculate the mean absorbance and %CV for the entire plate, for the interior wells (wells not on the outer perimeter), and for the edge wells separately.
- Validation Criterion: A successful sealing method will demonstrate a %CV of <5% across the entire plate and no statistically significant difference (p>0.05 by t-test) between the mean absorbance of the edge and interior wells.
Table 2: Quantitative Data Analysis from Environmental Control Experiments
| Experiment | Test Condition | Mean Signal (OD) | Background (OD) | Signal-to-Noise Ratio | Intra-Assay %CV | Calculated Conc. of Std. (pg/mL) |
|---|---|---|---|---|---|---|
| Temp. Profiling | 4°C Incubation | 1.25 | 0.08 | 15.6 | 4.5% | 245 |
| 22°C Incubation | 1.45 | 0.09 | 16.1 | 5.1% | 250 | |
| 37°C Incubation | 1.65 | 0.15 | 11.0 | 12.8% | 270 | |
| 40°C Incubation | 1.20 | 0.25 | 4.8 | 18.5% | 310 | |
| Edge Effect Mapping | Adhesive Sealer A | 0.75 | - | - | 3.2% | - |
| Silicone Cap Mat B | 0.76 | - | - | 4.8% | - | |
| No Seal | 1.10 | - | - | 25.5% | - |
The Scientist's Toolkit: Research Reagent Solutions
The consistent performance of the protocols above relies on the use of specific, high-quality materials. The following table details essential reagents and tools for implementing robust environmental controls.
Table 3: Essential Reagents and Tools for Environmental Control
| Item | Function/Description | Optimization Tip |
|---|---|---|
| High-Binding Polystyrene Plates | Solid phase for immobilizing capture antibody; plate composition can influence protein binding and edge effects [55]. | Test plates from different manufacturers; use plates from the same lot for a single study to minimize variability [55]. |
| Validated Antibody Pairs | Matched capture and detection antibodies that bind to non-overlapping epitopes of the target antigen [7] [52]. | Use affinity-purified antibodies. Titrate concentrations using a checkerboard assay for optimal signal-to-noise [7]. |
| Blocking Buffers | Inert protein solutions (e.g., BSA, non-fat dry milk) or commercial proprietary solutions used to coat unused binding sites on the plate [8] [52]. | Test different blockers and concentrations. If background is high, try adding a small amount of blocker to the wash buffer [54]. |
| Precision Incubators | Equipment for maintaining consistent temperature across all wells during incubations [52]. | Avoid stacking plates during incubation to ensure even heat distribution. Use an incubator with a built-in fan for circulation [54]. |
| Adhesive Plate Sealers | Films to prevent evaporation and cross-contamination during incubations [54]. | Use a fresh sealer for each incubation step. Press firmly around the entire plate rim to ensure a complete seal [54]. |
| Automated Plate Washer | Instrument for consistent and thorough washing across all wells to remove unbound reagents [52] [54]. | If washing manually, ensure consistent technique. Include a 30-second soak step between washes to improve elution of unbound material [54]. |
Standard Operating Procedure for Mitigation
Based on the experimental data, the following SOP steps are recommended for routine integration into any ELISA workflow to control for these variables.
- Temperature Uniformity: Pre-warm all reagents (except enzyme conjugates, if recommended otherwise) and the plate to the assay temperature before beginning. Use a calibrated, forced-air incubator and avoid stacking plates to ensure uniform thermal distribution [52] [54].
- Evaporation Prevention: Seal plates with a high-quality, adhesive seal during every incubation step. For extended incubations (>1 hour), place a humidifying tray containing clean water in the bottom of the incubator to maintain ambient humidity [54].
- Edge Effect Neutralization: When laying out the plate, place all buffer-only blanks, standards, and critical samples in both edge and interior wells. This allows for direct monitoring and mathematical correction of any edge-related bias if it occurs. Alternatively, fill all unused wells with a solution matching the sample matrix (e.g., PBS + 1% BSA) to create a uniform thermal mass across the entire plate [54].
- Washing Consistency: Use an automated plate washer where possible. If manual washing is necessary, use a multichannel pipette and a consistent, reproducible technique, ensuring wells are filled completely and aspirated thoroughly without touching the well bottom [52] [54].
The workflow for implementing these controls is summarized in the following diagram.
Ensuring Reliability: Assay Validation, Standardization, and Technology Comparison
Within the broader context of Enzyme-Linked Immunosorbent Assay (ELISA) protocol optimization research, the validation of analytical methods stands as a critical pillar for ensuring data reliability and reproducibility. For researchers, scientists, and drug development professionals, demonstrating that an ELISA performs consistently and reliably for its intended purpose is not merely a best practice but a regulatory necessity, particularly in pharmaceutical development where assays must comply with guidelines from agencies like the U.S. Food and Drug Administration (FDA) [56]. This document details the core experimental protocols and application notes for establishing four key validation parameters—Accuracy, Precision, Sensitivity, and Specificity—which form the foundation of a robust ELISA method. Thorough validation provides confidence in results, upholds the safety and efficacy of drug products, and is an indispensable component of any thesis focused on ELISA optimization [56] [57].
Core Validation Parameters: Definitions and Experimental Protocols
The following section defines each critical validation parameter and outlines the standardized experimental methodologies required to quantify them. These protocols are designed to be integrated into a comprehensive ELISA optimization workflow.
Accuracy
- Definition: Accuracy measures the closeness of agreement between the value found in a sample and the value accepted as either a conventional true value or an accepted reference value. It indicates how close your results are to the true concentration of the analyte [57] [58].
- Importance: An accurate ELISA produces reliable and valid data, which is fundamental for informed decision-making in both diagnostic and drug development settings [58].
- Experimental Protocol: Spike and Recovery
The following procedure evaluates the impact of the sample matrix on accuracy and determines the percentage recovery of a known amount of analyte [8].
- Preparation: Prepare a sample matrix known to be free of the target analyte (e.g., stripped serum or a suitable buffer).
- Spiking: Spike a known, pure quantity of the analyte (the "spike") into the sample matrix. The concentration of the spiked analyte should fall within the working range of the ELISA.
- Control: Prepare a parallel sample by spiking the same known quantity of the analyte into the standard diluent used to generate the calibration curve.
- Analysis: Run both the spiked matrix sample and the spiked diluent control through the ELISA in replicate (typically n≥5).
- Calculation: Calculate the percentage recovery for the spiked matrix sample using the formula:
Recovery % = (Measured Concentration in Spiked Matrix / Measured Concentration in Spiked Diluent) × 100 - Interpretation: Ideally, the recovery should be close to 100%. Consistent deviations (e.g., consistently low or high recovery) indicate a matrix effect that interferes with the accuracy of the assay and must be addressed, often by modifying the sample diluent [8].
Precision
- Definition: Precision is the measure of the degree of scatter between a series of measurements obtained from multiple sampling of the same homogeneous sample under prescribed conditions. It is an indicator of the assay's reproducibility and is usually expressed as the coefficient of variation (%CV) [58].
- Importance: A highly precise assay yields consistent results over time and across different operators, which is vital for tracking biological changes or treatment effects reliably [57] [58].
- Experimental Protocol: Repeatability and Intermediate Precision
Precision is assessed at multiple levels to ensure consistency [57].
- Sample Preparation: Prepare three different pools of samples with analyte concentrations representing low, medium, and high levels within the dynamic range of the assay.
- Repeatability (Intra-assay Precision): Analyze each sample pool multiple times (e.g., n=8-10) within a single assay run by the same operator using the same reagents and equipment. Calculate the mean, standard deviation (SD), and %CV for each pool.
- Intermediate Precision (Inter-assay Precision): Analyze the same three sample pools across multiple independent assay runs (e.g., on different days, with different operators, or using different reagent lots). A minimum of three separate runs is recommended.
- Calculation: For both intra- and inter-assay measurements, calculate the %CV as:
%CV = (Standard Deviation / Mean) × 100. - Interpretation: The obtained %CV values are evaluated against pre-defined acceptance criteria (e.g., %CV < 15% for biological samples). Meeting these criteria confirms the assay's robustness against minor operational variations [56].
Sensitivity
- Definition: Sensitivity refers to the lowest concentration of an analyte that an assay can reliably differentiate from zero. It represents the detection limit of the assay and is crucial for detecting low-abundance targets [58].
- Importance: High sensitivity is paramount for the early diagnosis of diseases (e.g., detecting low levels of cancer biomarkers), quantifying analytes in limited sample volumes, and monitoring minute biological changes in research [23] [58].
- Experimental Protocol: Limit of Detection (LOD) and Limit of Quantification (LOQ)
This protocol establishes the lowest measurable concentrations [58].
- Sample Preparation: Prepare a minimum of 16-20 replicates of a blank sample (a sample containing all assay components except the analyte) and a series of low-concentration calibrators near the expected detection limit.
- Analysis: Measure all replicates in a single assay.
- LOD Calculation: The LOD is typically calculated as the mean signal of the blank plus 2 or 3 standard deviations (SD). The concentration corresponding to this signal is the LOD.
LOD = Mean_blank + 3 × SD_blank - LOQ Calculation: The LOQ is the lowest concentration that can be measured with acceptable precision and accuracy. It is often defined as the mean signal of the blank plus 10 standard deviations or as the lowest concentration on the standard curve that can be measured with a %CV ≤ 20%.
LOQ = Mean_blank + 10 × SD_blank - Interpretation: The LOD and LOQ define the lower boundary of the assay's working range. An assay with a lower LOD/LOQ possesses higher sensitivity.
Specificity
- Definition: Specificity is the ability of the assay to measure the analyte unequivocally in the presence of other components that may be expected to be present in the sample, such as structurally similar molecules, metabolites, or other matrix components [57] [58].
- Importance: High specificity ensures that the measured signal is generated solely by the target analyte and not by cross-reacting substances, which is critical for accurate diagnosis and valid research conclusions [58].
- Experimental Protocol: Cross-Reactivity and Interference Testing
- Cross-Reactivity Assessment: Test potentially cross-reacting substances (e.g., analogs, metabolites, related proteins) by spiking them individually into the assay at physiologically relevant high concentrations. The assay's response to these substances is compared to its response to the actual analyte.
- Calculation: Calculate the percentage cross-reactivity as:
Cross-reactivity % = (Concentration of Analyte / Concentration of Cross-reactant) × 100, where the concentrations are those that produce the same assay response (e.g., at 50% binding). - Interference Testing: To assess matrix effects, perform a "parallelism" experiment. Serially dilute a sample known to contain a high endogenous level of the analyte and plot the measured concentration against the dilution factor. The curve should be parallel to the standard curve.
- Interpretation: A specific assay will show low cross-reactivity (<1-5% for critical interferents) and demonstrate linearity and parallelism, confirming that the sample matrix does not adversely affect analyte detection [8].
Data Presentation and Analysis
Table 1: Summary of Key ELISA Validation Parameters, their Definitions, and Target Acceptance Criteria.
| Parameter | Definition | Typical Experimental Method | Common Acceptance Criteria |
|---|---|---|---|
| Accuracy | Closeness to the true value [58] | Spike and Recovery [8] | Recovery of 80-120% [57] |
| Precision | Closeness of repeated measurements [58] | Repeatability (Intra-assay) & Intermediate Precision (Inter-assay) [57] | %CV < 10-15% [56] [57] |
| Sensitivity | Lowest detectable concentration [58] | Limit of Detection (LOD) / Limit of Quantification (LOQ) [58] | Signal-to-Noise ratio ≥ 2-3 (LOD) [58] |
| Specificity | Ability to detect only the target [57] | Cross-reactivity & Parallelism Testing [8] | Cross-reactivity < 1-5%; Parallelism %CV < 20% [8] |
Experimental Workflow for ELISA Validation
The following diagram illustrates the logical sequence and relationships between the key experiments in a comprehensive ELISA validation workflow.
The Scientist's Toolkit: Essential Research Reagent Solutions
The reliability of ELISA validation is contingent upon the quality and consistency of the reagents used. The following table details the essential materials and their functions.
Table 2: Key Research Reagent Solutions for ELISA Validation Experiments.
| Reagent / Material | Function in Validation | Key Considerations |
|---|---|---|
| Coating Antibody | Captures the target analyte onto the solid phase [7] | Affinity-purified antibodies are recommended for optimal signal-to-noise ratio; concentration must be optimized (e.g., 1-12 µg/mL) [7]. |
| Detection Antibody | Binds to the captured analyte; conjugated to an enzyme for detection [7] | Must be specific for a different epitope than the capture antibody (matched pair); concentration requires optimization (e.g., 0.5-5 µg/mL) [7]. |
| Reference Standard | Pure analyte used to generate the calibration curve [8] | Critical for accuracy; should be of known purity and identity. Use the same batch for all validation experiments to avoid variability [8]. |
| Sample Diluent | Matrix used to dilute samples and standards [7] | Should match the sample matrix as closely as possible to minimize matrix effects in spike/recovery and parallelism experiments [7] [8]. |
| Blocking Buffer | Prevents non-specific binding to the plate [7] | Essential for achieving high specificity and low background. Different proteins (e.g., BSA, casein) should be tested during optimization [7]. |
| Enzyme Conjugate | Catalyzes the conversion of a substrate to a detectable product [4] | Common enzymes are Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP). Concentration must be optimized for the chosen substrate (e.g., 20-200 ng/mL for HRP colorimetric) [7] [4]. |
| Control Samples | Positive, negative, and blank controls [8] | Used in every run to monitor assay performance, precision, and to validate results. Positive controls confirm expected signal; negative controls assess background [8]. |
The rigorous validation of an ELISA protocol through the systematic assessment of accuracy, precision, sensitivity, and specificity is a non-negotiable standard in scientific research and drug development. The experimental protocols and application notes detailed herein provide a clear roadmap for researchers to establish these parameters, ensuring that the data generated is reliable, reproducible, and fit for its intended purpose. As the field advances with next-generation ELISA technologies offering enhanced multiplexing and sensitivity, the fundamental principles of method validation remain constant, forming the bedrock of scientific integrity and the advancement of knowledge in biomedicine [56] [23].
In the context of enzyme-linked immunosorbent assay (ELISA) protocol optimization, demonstrating that an assay is fit-for-purpose is a fundamental requirement in research and drug development. Two critical experimental procedures used for this validation are spike-and-recovery and dilution linearity. These experiments are essential for assessing the accuracy of an ELISA and identifying whether components in a sample matrix interfere with the detection and quantification of the target analyte [8] [59]. Spike-and-recovery experiments determine if the sample matrix affects the assay's ability to detect a known amount of analyte, while dilution linearity tests evaluate the precision of results across different sample dilutions, ensuring the assay's response is consistent and proportional [59] [60]. For researchers and scientists, particularly in regulated environments, successfully performing these experiments is crucial for generating reliable, high-quality data and qualifying an ELISA for use in critical applications, such as process development and product purity testing in biopharmaceuticals [61].
The Principle and Purpose of Spike-and-Recovery Experiments
Core Concept and Importance
Spike-and-recovery is a fundamental validation experiment designed to diagnose matrix effects in immunoassays. The core principle involves adding ("spiking") a known quantity of the purified analyte into the natural sample matrix and then measuring the amount recovered by the ELISA [59] [61]. The goal is to achieve identical assay responses for a given amount of analyte, whether it is in the standard diluent (used for the standard curve) or the sample matrix (e.g., serum, cell culture supernatant, or drug substance) [59]. When the recovery differs, it indicates that components within the sample matrix—such as high or low pH, high protein or salt concentration, detergents, or organic solvents—are interfering with the antibody-antigen interaction or the enzymatic detection system [61]. This interference can lead to either an overestimation (over-recovery) or, more commonly, an underestimation (under-recovery) of the true analyte concentration, compromising the accuracy of the entire study [61].
When to Perform Spike-and-Recovery
Spike-and-recovery analysis is not a one-time experiment. It should be performed:
- For each unique sample type (e.g., harvest cell culture fluid, in-process samples, final drug product) with a distinct matrix composition [61].
- After establishing the Minimum Required Dilution (MRD) through dilution linearity studies, as the sample will be tested at this dilution to minimize matrix effects [60] [61].
- Whenever there is a change in the manufacturing process that could alter the sample matrix [61].
Protocol for Spike-and-Recovery Experiments
Experimental Workflow
The following diagram outlines the key steps in a spike-and-recovery experiment.
Step-by-Step Methodology
- Prepare the Sample Matrix: Begin with the sample (e.g., final product, serum) at its predetermined Minimum Required Dilution (MRD) to minimize matrix interference [61].
- Spike the Sample: Introduce a known amount of purified analyte (the "spike") into the diluted sample matrix. The spike should be prepared at 3-4 concentration levels covering the analytical range of the ELISA, with the lowest concentration being at least two times the Limit of Quantitation (LOQ) of the assay [61]. For example, to achieve a final spike concentration of 20 ng/mL, you might add 1 part of a 100 ng/mL standard into 4 parts of the neat sample [61].
- Prepare the Control: In parallel, spike the same known amount of analyte into the standard diluent used to generate the standard curve. This serves as the control, representing the ideal recovery scenario without matrix effects [59].
- Run the ELISA: Assay both the spiked sample matrix and the spiked control diluent in the ELISA following the established protocol. It is also essential to run a non-spiked sample (sample matrix + assay diluent instead of spike) to determine the background level of endogenous analyte [61].
- Calculate Recovery: After measuring the optical density and interpolating concentrations from the standard curve, calculate the percentage recovery using the formula below. Regulatory guidelines from ICH, FDA, and EMA typically consider recovery values within 75% to 125% as acceptable [61].
Table 1: Sample Data for Spike-and-Recovery Calculation
| Sample Component | HCP Measured (ng/mL) | Calculation |
|---|---|---|
| Sample + "Zero Standard" (Endogenous HCP) | 6 | N/A |
| Sample + "100 ng/mL Standard" (Total HCP) | 25 | N/A |
| Recovered Spike | 19 | (25 - 6) |
| Expected Spike | 20 | (From spike) |
| % Recovery | 95% | (19 / 20) × 100% |
The Principle and Purpose of Dilution Linearity Experiments
Core Concept and Importance
Dilution linearity, or linearity-of-dilution, assesses whether the measured concentration of an analyte in a sample is consistent and proportional across a series of dilutions [59] [60]. The purpose of this experiment is to establish the dose-response curve and the full quantitative range of the assay for the specific sample type [60]. A key outcome is determining the Minimum Required Dilution (MRD), which is the lowest dilution at which the sample can be analyzed while still yielding accurate results [60]. When linearity is demonstrated, it confirms that the condition of antibody excess is met for all analytes in the sample, meaning the assay antibodies are not saturated and can accurately quantify the target [60]. Poor linearity, where the dilution-corrected concentration changes significantly with dilution, indicates potential issues such as matrix interference, the "high-dose hook effect" from antibody saturation, or the presence of "hitchhiker proteins" that interact with the product [60].
Protocol for Dilution Linearity Experiments
Experimental Workflow
The workflow for establishing dilution linearity is methodical, as shown below.
Step-by-Step Methodology
- Select Sample and Diluent: Choose a sample with a suspected analyte concentration above the assay's limit of quantification. Use an appropriate assay diluent, ideally the same one used to prepare the kit standards [60] [62].
- Perform Serial Dilutions: Create a series of doubling dilutions (e.g., neat, 1:2, 1:4, 1:8, etc.) of the sample in the chosen diluent. The dilution series should extend to a point where the concentration falls below the lower limit of the standard curve [59] [60].
- Run the ELISA: Assay all dilutions in the ELISA alongside the standard curve.
- Calculate and Analyze Data: For each dilution, calculate the dilution-corrected concentration (Observed Concentration × Dilution Factor). Analyze the corrected values to identify the MRD. The acceptable range for dilution linearity is typically defined as a variation of no more than ±20% between consecutive doubling dilutions, and the observed concentration (before correction) should be above two times the assay's LOQ [60].
Table 2: Example of Dilution Linearity Data Analysis for MRD Determination
| Sample Dilution | Dilution Corrected Value (ng/mL) | % Change from Previous Dilution | Meets Criteria? |
|---|---|---|---|
| Neat (undiluted) | 146 | N/A | No |
| 1:2 | 233 | 60% | No |
| 1:4 | 312 | 34% | No |
| 1:8 | 361 | 16% | Yes |
| 1:16 | 356 | 1% | Yes |
| 1:32 | 370 | 4% | Yes |
| 1:64 | (Not calculated, <2x LOQ) | N/A | No |
From the data in Table 2, the MRD is determined to be 1:8, as this is the most concentrated dilution where the dilution-corrected values stabilize and show minimal variation (<±20%) in subsequent dilutions that are above the LOQ. The reported HCP concentration for this sample would be the average of the values from the 1:8, 1:16, and 1:32 dilutions [60].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of spike-and-recovery and dilution linearity experiments requires careful selection of reagents and materials to ensure data integrity and reproducibility.
Table 3: Essential Materials for Assay Validation Experiments
| Item | Function & Importance | Key Considerations |
|---|---|---|
| Purified Analyte Standard | Serves as the "spike" and is used to generate the standard curve for quantification. | Should be highly pure and well-characterized. The standard used for spiking must be identical to the one used for the standard curve [59]. |
| Appropriate Diluent Buffers | Used to dilute samples, standards, and reagents. Critical for maintaining pH and ionic strength. | The standard diluent should ideally match the sample matrix as closely as possible (e.g., using culture medium for culture supernatant samples) to minimize matrix effects [59] [7]. |
| Matched Antibody Pairs | The capture and detection antibodies are the core of a sandwich ELISA, defining its specificity. | Antibodies should be affinity-purified and used at optimized concentrations to achieve a high signal-to-noise ratio [7]. |
| Microplates and Readers | 96-well microplates serve as the solid phase; the plate reader quantifies the colorimetric signal. | Use plates with high protein-binding capacity. The plate reader should be capable of reading at the appropriate wavelength (e.g., 450 nm for TMB substrate) [4]. |
| Precision Pipettes and Liquid Handling | Critical for accurate serial dilutions and reagent dispensing, which directly impact data variability. | Automated liquid handlers can significantly improve accuracy, reproducibility, and throughput while reducing human error [63]. |
Troubleshooting Common Issues
Even with a well-designed experiment, issues can arise. The table below outlines common problems and their potential solutions.
Table 4: Troubleshooting Guide for Poor Assay Performance
| Observed Issue | Potential Cause | Corrective Actions |
|---|---|---|
| Poor Spike-and-Recovery (<75% or >125%) | Sample matrix components interfere with antibody binding or enzyme activity [59] [61]. | - Further dilute the sample to reduce matrix interference [59] [61].- Alter the standard diluent to more closely match the sample matrix (e.g., by adding a carrier protein like BSA) [59] [7]. |
| Poor Dilution Linearity | Antibody saturation for one or more analytes ("hook effect") or matrix interference at high concentrations [60]. | - Increase the sample dilution to ensure measurements are taken within the antibody excess region [60].- Re-optimize antibody concentrations if developing an ELISA from scratch [8] [7]. |
| High Background Signal | Non-specific binding of detection antibodies or insufficient washing [62]. | - Optimize the composition and concentration of the blocking buffer [8] [7].- Increase the number or duration of wash steps [8] [62]. |
| Inconsistent Replicates | Manual pipetting errors, improper mixing, or uneven coating/incubation [63]. | - Use automated liquid handling systems for dispensing and dilutions [63].- Ensure consistent and thorough mixing of all reagents and samples [62]. |
Spike-and-recovery and dilution linearity experiments are non-negotiable components of a robust ELISA optimization strategy. They provide the empirical data needed to confirm that an assay delivers accurate and precise results for specific sample matrices, a cornerstone of reliable scientific research and drug development. By following the detailed protocols outlined in this document—employing a systematic workflow, utilizing essential reagents, and applying logical troubleshooting—scientists can confidently qualify their ELISA methods, ensure regulatory compliance, and generate data that truly reflects the biological reality of their samples.
The Enzyme-Linked Immunosorbent Assay (ELISA) remains one of the most widely used techniques in biomedical research and clinical diagnostics for detecting and quantifying peptides, proteins, antibodies, and hormones [4] [13]. Despite its widespread adoption and apparent simplicity, achieving consistent, reproducible results across different laboratories presents significant challenges due to the multi-step nature of the technique and numerous variables that require careful control. Reproducibility is the cornerstone of scientific integrity, enabling reliable comparison of data between research groups, longitudinal studies, and multi-center clinical trials [64].
The fundamental principle of ELISA relies on the specific antigen-antibody interaction, where the target molecule is immobilized on a solid surface (typically a microplate) and detected using an antibody linked to a reporter enzyme that generates a measurable signal upon substrate addition [4] [13]. While this core principle remains constant, variations in protocol execution, reagent selection, and analytical approaches can introduce substantial inter-laboratory variability. This application note provides a comprehensive framework for standardizing ELISA protocols to enhance reproducibility, with specific methodologies for validation, optimization, and implementation of consistent practices across laboratories.
Foundational ELISA Validation Parameters
Robust ELISA performance requires systematic validation of key parameters that collectively determine the assay's reliability and transferability between laboratories. The following parameters form the essential foundation for reproducible inter-laboratory results.
Table 1: Essential Validation Parameters for Reproducible ELISA Results
| Parameter | Definition | Acceptance Criteria | Impact on Reproducibility |
|---|---|---|---|
| Specificity | Ability to accurately measure target analyte without cross-reactivity | Minimal interference from related proteins or matrix components | Ensures different laboratories measure the same target |
| Sensitivity | Lowest detectable concentration with statistical significance | Limit of Detection (LOD): Mean background + 2SD [64] | Determates lower quantification limits across labs |
| Functional Sensitivity | Lowest concentration that can be reliably quantified | Lower Limit of Quantification (LLOQ): CV <20% [64] | Defines working range for comparative studies |
| Accuracy/Recovery | Agreement between measured and true values | 80-120% recovery of spiked analyte [64] | Confirms minimal matrix interference between lab settings |
| Precision | Agreement between replicate measurements | Within-run CV <10%; Between-run CV <15% [64] | Critical for longitudinal and multi-center studies |
| Parallelism | Equivalent detection of endogenous and standard curve analyte | %CV <20% across dilutions [8] | Ensures standard curve accurately reflects sample matrix |
Establishing Sensitivity Parameters
Sensitivity validation requires calculating both the Limit of Detection (LOD) and Lower Limit of Quantification (LLOQ). The LOD represents the lowest concentration that can be detected with statistical significance, typically calculated as the mean background signal plus two standard deviations [64]. The LLOQ represents the lowest concentration that can be quantitatively measured with acceptable precision and accuracy, generally defined as the concentration where the coefficient of variation (CV) is less than 20% [64]. These parameters must be established using the same matrix as experimental samples to account for matrix effects.
Precision and Accuracy Assessment
Precision validation encompasses both within-run (intra-assay) and between-run (inter-assay) measurements [64]. Within-run precision is evaluated by testing multiple replicates of one sample in a single assay run, while between-run precision assesses variation across different operators, days, and reagent lots. Accuracy is typically validated through spike-and-recovery experiments, where known quantities of purified analyte are added to sample matrices and recovery percentages are calculated [64] [65]. Ideal recovery falls between 80-120%, indicating minimal matrix interference.
Standardized Experimental Protocols
Protocol 1: Comprehensive Assay Validation
Purpose: To establish performance characteristics of an ELISA method for inter-laboratory use.
Materials:
- ELISA kits or components (matched antibody pairs, coated plates)
- Reference standards calibrated to international standards (e.g., NIBSC, WHO) [64]
- Appropriate biological matrices (serum, plasma, cell lysate)
- Microplate reader capable of absorbance measurement at appropriate wavelengths (e.g., 450nm)
- Precision pipettes, including multichannel pipettes
- Plate washer or washing buffer system
Procedure:
- Precision Testing: Run three levels of quality control samples (low, medium, high) with 6 replicates each in the same run for within-run precision. Repeat this process over 6 separate days for between-run precision [64].
- Linearity and Parallelism: Prepare serial dilutions of a sample with high endogenous analyte and a spiked sample. Analyze diluted samples against the standard curve [8].
- Spike-and-Recovery: Add known concentrations of purified analyte to at least 5 different sample matrices. Calculate percentage recovery by comparing measured values to expected values [64] [65].
- Specificity Assessment: Test cross-reactivity with structurally similar molecules and potential interfering substances (e.g., hemolyzed, lipemic, or icteric samples).
Data Analysis:
- Calculate mean, standard deviation (SD), and coefficient of variation (CV%) for precision samples.
- For linearity, perform linear regression analysis and calculate the coefficient of determination (R²).
- For recovery, calculate: (Measured Concentration / Expected Concentration) × 100.
Protocol 2: Checkerboard Titration for Assay Optimization
Purpose: To simultaneously optimize multiple assay parameters using a systematic titration approach.
Materials:
- Coating antibody or antigen at various concentrations
- Detection antibody at various concentrations
- Blocking buffers (e.g., BSA, casein, non-fat dry milk)
- Coating buffers (carbonate-bicarbonate buffer, pH 9.4 or PBS, pH 7.4) [4] [7]
Procedure:
- Prepare a 96-well plate map with capture reagent concentrations varying across rows and detection reagent concentrations varying down columns [8] [7].
- Coat wells with different concentrations of capture antibody (typically 1-15 μg/mL depending on antibody source) [7].
- Block wells with different blocking solutions (e.g., 1-5% BSA, casein, or commercial blockers).
- Add standard concentration of analyte followed by detection antibodies at varying concentrations (typically 0.5-10 μg/mL) [7].
- Develop with substrate and measure signal.
- Identify the combination that provides the highest signal-to-background ratio.
Table 2: Recommended Antibody Concentrations for ELISA Optimization
| Antibody Source | Coating Antibody Range | Detection Antibody Range |
|---|---|---|
| Polyclonal serum | 5–15 μg/mL | 1–10 μg/mL |
| Crude ascites | 5–15 μg/mL | 1–10 μg/mL |
| Affinity-purified polyclonal | 1–12 μg/mL | 0.5–5 μg/mL |
| Affinity-purified monoclonal | 1–12 μg/mL | 0.5–5 μg/mL |
Protocol 3: Matrix Interference and Parallelism Assessment
Purpose: To evaluate and mitigate matrix effects that compromise inter-laboratory comparability.
Procedure:
- Prepare a pooled sample matrix with high endogenous analyte or spike a pool with recombinant protein.
- Create a series of serial dilutions (e.g., 1:2, 1:4, 1:8, 1:16) using the appropriate sample diluent.
- Run all dilutions in the same assay alongside the standard curve.
- Plot measured concentrations against expected concentrations (based on dilution factors).
- Perform linear regression analysis to evaluate parallelism.
Interpretation: A slope close to 1.0 and high R² value indicate minimal matrix effects and proper parallelism [8] [64]. Significant deviations suggest matrix interference requiring modification of sample diluent or dilution factor.
Controls and Standards for Reproducibility
Implementing a comprehensive control strategy is essential for monitoring assay performance and ensuring reproducibility across laboratories and over time.
Table 3: Essential Controls for Reproducible ELISA Operations
| Control Type | Purpose | Composition | Interpretation |
|---|---|---|---|
| Blank Control | Measure background from plastic/buffer | Wells with assay buffer only [65] | Signal should be minimal |
| S0 Control (Zero Standard) | Determine background in absence of analyte | All assay components except analyte [65] | Defines baseline for standard curve |
| Non-Specific Binding (NSB) Control | Assess nonspecific binding in competitive ELISA | No capture antibody, with conjugate [65] | Identifies conjugate binding issues |
| Positive Control | Verify assay functionality | Known amount of target analyte | Should fall within expected range |
| Standard Curve | Quantification reference | Serial dilutions of known analyte | R² > 0.99 for optimal assays |
Implementation of Standard Operating Procedures
Reagent Standardization
Consistent reagent preparation and storage are fundamental to inter-laboratory reproducibility:
- Coating Buffer: Standardize on carbonate-bicarbonate buffer (pH 9.4) or PBS (pH 7.4) with documented preparation protocols [4] [7].
- Wash Buffer: Use PBS with 0.05% Tween-20 with defined pH (7.4) and storage conditions.
- Blocking Buffer: Document specific blocking agents (BSA, casein, etc.) with precise concentrations and incubation parameters.
- Antibody Standards: Utilize international reference standards where available [64]. For custom assays, establish in-house reference preparations with detailed characterization.
Equipment Calibration and Maintenance
Regular calibration of key equipment ensures measurement consistency:
- Pipettes: Perform quarterly gravimetric calibration with documentation.
- Microplate Readers: Monthly verification using neutral density filters or standardized chromogenic solutions.
- Incubators: Continuous temperature monitoring with calibrated probes.
- Plate Washers: Regular inspection of dispense and aspiration volumes and patterns.
Data Analysis and Reporting Standards
Standardized approaches to data analysis prevent interpretation variability:
- Standard Curve Fitting: Use 4-parameter logistic (4PL) regression for most immunoassays [8].
- Outlier Identification: Establish predefined criteria for excluding data points (e.g., CV > 20% between replicates).
- Minimum Required Information: Include all critical assay parameters in publications: antibody lots, incubation times/temperatures, plate reader settings, and validation data.
Workflow Visualization
Figure 1: ELISA Validation and Standardization Workflow. This diagram illustrates the systematic process for developing and validating reproducible ELISA methods.
Figure 2: ELISA Format Selection and Workflows. Different ELISA formats require specific standardization approaches for inter-laboratory consistency.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Research Reagent Solutions for Reproducible ELISA
| Reagent Category | Specific Examples | Function | Standardization Guidelines |
|---|---|---|---|
| Solid Phase | 96-well microplates (polystyrene, polyvinyl) | Analyte immobilization | High binding capacity (>400ng/cm²), low well-to-well CV (<5%) [13] |
| Coating Buffers | Carbonate-bicarbonate (pH 9.4), PBS (pH 7.4) | Facilitate passive adsorption | Document pH precisely; filter sterilize for stability |
| Blocking Agents | BSA, casein, non-fat dry milk, commercial blockers | Cover unsaturated binding sites | Standardize concentration (1-5%) and incubation time |
| Detection Enzymes | Horseradish peroxidase (HRP), Alkaline phosphatase (AP) | Signal generation | Standardize conjugation procedures and concentrations [4] [7] |
| Antibody Systems | Matched antibody pairs, primary-secondary systems | Target recognition and detection | Use affinity-purified antibodies; validate cross-reactivity [7] |
| Reference Standards | International standards (NIBSC, WHO), in-house references | Quantification calibration | Establish master stock with proper storage (-80°C) [64] |
Achieving consistent inter-laboratory reproducibility with ELISA requires meticulous attention to validation, standardization, and documentation. By implementing the systematic approaches outlined in this application note—comprehensive assay validation, standardized operating procedures, appropriate controls, and consistent data analysis—researchers can significantly enhance the reliability and comparability of their ELISA data. The standardized protocols provided herein serve as a foundation for establishing robust ELISA methods that yield consistent results across different laboratory settings, thereby strengthening research outcomes and facilitating collaborative science.
The enzyme-linked immunosorbent assay (ELISA) has long been the gold standard for protein biomarker detection and quantification in clinical diagnostics and drug development [66] [13]. Its robustness, specificity, and accessibility have made it a fundamental tool in research laboratories worldwide. However, the era of precision medicine demands more rigorous biomarker validation methods with enhanced sensitivity, broader dynamic range, and multiplexing capabilities [66]. While traditional ELISA remains valuable, advanced technologies such as Meso Scale Discovery (MSD) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) now offer superior performance characteristics for challenging applications. This application note provides a detailed comparison of these technologies within the context of ELISA protocol optimization research, offering structured experimental protocols and performance data to guide researchers in selecting appropriate platforms for their specific needs.
Technology Comparison and Performance Metrics
Key Advantages and Limitations
Traditional ELISA operates on the principle of antibody-antigen interaction, where a target macromolecule is immobilized on a solid surface and complexed with an antibody linked to a reporter enzyme [13]. The most common format is the sandwich ELISA, which uses two antibodies targeting different epitopes on the antigen for enhanced specificity [13]. Despite its widespread use, ELISA faces limitations including a relatively narrow dynamic range (typically 1-2 logs), substantial sample volume requirements (50-100 μL per analyte), and inability to multiplex without running separate assays [67] [68]. Additionally, its performance is highly dependent on antibody quality, with potential for cross-reactivity and limited detection of low-abundance biomarkers [66] [69].
The Meso Scale Discovery (MSD) platform represents an advanced electrochemiluminescence-based immunoassay technology. Unlike ELISA, which relies on colorimetric or chemiluminescent signals from enzyme-substrate reactions, MSD utilizes electrode-lined microplates and ruthenium-labeled detection antibodies [68]. When electrical stimulation is applied, the ruthenium label emits light, which is detected by the instrument's camera [68]. This methodology provides up to 100 times greater sensitivity than traditional ELISA and a broader dynamic range (3-4+ logs) [66] [67]. MSD's multiplexing capability allows simultaneous measurement of up to 10 analytes from a single small sample volume (10-25 μL) [67], significantly enhancing efficiency in biomarker research, especially when dealing with complex diseases or therapeutic responses [66].
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) offers a fundamentally different approach, based on physical separation and mass analysis rather than antibody binding [69]. This technique combines high-performance liquid chromatography with tandem mass spectrometry to separate, identify, and quantify biomolecules with unparalleled specificity [69] [70]. LC-MS/MS provides direct, molecule-by-molecule analysis, enabling identification of specific protein isoforms and post-translational modifications that often remain undetected by immunoassays [69] [71]. With sensitivity reaching the atto- to femtomolar range and a dynamic range of 4-6 logs, LC-MS/MS surpasses both ELISA and MSD in detection capabilities [41] [68]. Furthermore, it allows for multiplexing hundreds to thousands of proteins in a single run [66], making it particularly valuable for comprehensive biomarker discovery and validation.
Quantitative Performance Comparison
Table 1: Analytical Performance Metrics of ELISA, MSD, and LC-MS/MS
| Parameter | Traditional ELISA | MSD Platform | LC-MS/MS |
|---|---|---|---|
| Sensitivity | ~1-2 logs [68] | ~3-4+ logs [68], Up to 100x more sensitive than ELISA [66] | ~4-6 logs [68], Attomolar to femtomolar range [41] |
| Dynamic Range | 1-2 logs [67] [68] | 3-4+ logs [67] [68] | 4-6 logs [68] |
| Sample Volume | 50-100 μL (per analyte) [67] | 10-25 μL (for up to 10 analytes) [67] | Varies by protocol, typically 10-200 μL [70] |
| Multiplexing Capability | Single analyte per well [67] [68] | Up to 10 analytes simultaneously [67] [68] | Hundreds to thousands of proteins [66] |
| Matrix Effects | Can be high, depending on assay [68] | Generally low [67] [68] | Generally low [68] |
| Throughput Time | Slow (multiple long incubations) [68] | Fast (simpler protocols, fewer wash steps) [67] [68] | Fast analysis, but sample preparation can be lengthy [70] |
Table 2: Economic and Practical Considerations
| Consideration | Traditional ELISA | MSD Platform | LC-MS/MS |
|---|---|---|---|
| Equipment and Maintenance Costs | Low compared to other methods [68] | Medium compared to other methods [68] | Higher compared to other methods [68] |
| Reagent Costs | Relatively inexpensive [69] | Higher than ELISA, but cost-effective when multiplexing [66] | Expensive instrumentation and specialized expertise required [69] [70] |
| Operational Complexity | Simple, single-step assay [69] | Simple protocols, typically 1-3 wash steps [67] | Multistep, complex technique [69] |
| Example Cost per Sample | ~$61.53 (for 4 inflammatory biomarkers) [66] | ~$19.20 (for 4 biomarkers via multiplexing) [66] | Significant upfront investment, but cost-effective for large-scale analyses |
Experimental Protocols
MSD Immunoassay Protocol for Multiplex Biomarker Analysis
Principle: The MSD platform uses electrochemiluminescence detection with antibody-coated spots on multi-array plates [67]. Each spot contains a specific capture antibody, allowing simultaneous measurement of multiple analytes from a single sample [67].
Protocol Steps:
- Plate Preparation: Use MSD MULTI-ARRAY 96-well plates with pre-coated capture antibodies. Bring plates to room temperature before use.
- Sample and Standard Preparation:
- Prepare standards using recombinant proteins in the same matrix as samples.
- Dilute samples appropriately using the recommended diluent.
- Include quality controls at low, medium, and high concentrations.
- Assay Procedure:
- Add 25 μL of standards, controls, or samples to appropriate wells.
- Incubate plates for 2 hours with shaking at room temperature.
- Wash plates 3 times with PBS-Tween using an automated plate washer.
- Add 25 μL of detection antibody solution (ruthenium-conjugated detection antibodies) to each well.
- Incubate for 2 hours with shaking at room temperature.
- Wash plates 3 times as before.
- Add 150 μL of MSD Read Buffer T to each well.
- Data Acquisition:
- Read plates immediately using an MSD instrument.
- Measure light emission produced by electrical stimulation of ruthenium labels.
- Data Analysis:
- Generate standard curves for each analyte using 4-parameter logistic fit.
- Calculate sample concentrations from standard curves.
- Analyze data using MSD Discovery Workbench software.
Critical Steps for Optimization:
- Optimize antibody pairs for each biomarker through checkerboard titration.
- Validate assay performance in specific biological matrices.
- Determine optimal sample dilution to minimize matrix effects.
MSD Assay Workflow: This diagram illustrates the sequential steps for performing a multiplex immunoassay using the Meso Scale Discovery platform, from plate preparation to data analysis.
LC-MS/MS Protocol for Targeted Protein Quantification
Principle: LC-MS/MS combines liquid chromatography separation with tandem mass spectrometry detection, enabling highly specific identification and quantification of proteins based on mass-to-charge ratio [69] [70]. The protocol typically uses multiple reaction monitoring (MRM) for targeted quantification [72].
Protocol Steps:
- Sample Preparation:
- Add internal standard (e.g., isotopically labeled peptide, isodesmosine-13C3,15N1) to samples [70].
- For complex matrices, perform protein precipitation or solid-phase extraction.
- Digest proteins with trypsin (if quantifying proteins via surrogate peptides).
- Desalt samples using C18 columns or cartridges.
- Liquid Chromatography:
- Inject samples onto HPLC system with C18 column.
- Use gradient elution with mobile phase A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile).
- Typical gradient: 2-35% B over 30-60 minutes at flow rate of 0.2-0.4 mL/min.
- Mass Spectrometry Analysis:
- Use electrospray ionization in positive mode.
- Set MS parameters: capillary voltage 3.5 kV, source temperature 150°C, desolvation temperature 350°C.
- Configure MRM transitions for each target analyte and internal standard.
- For desmosine analysis: monitor transitions m/z 397.25→232.10 for desmosine and m/z 401.25→232.10 for isotopic internal standard [70].
- Data Processing:
- Integrate peak areas for each MRM transition.
- Calculate peak area ratios (analyte/internal standard).
- Generate calibration curves using linear regression with 1/x weighting.
- Determine sample concentrations from calibration curves.
Critical Steps for Optimization:
- Optimize MRM transitions for each target analyte.
- Validate selectivity, sensitivity, and linearity in biological matrices.
- Determine extraction efficiency and matrix effects.
LC-MS/MS Workflow: This diagram illustrates the key steps in LC-MS/MS analysis for targeted protein quantification, from sample preparation with internal standards to MRM-based quantitation.
Traditional ELISA Optimization Protocol
Principle: ELISA is based on the specific binding between an antigen and antibody, with detection enabled by an enzyme-labeled antibody and colorimetric, fluorescent, or chemiluminescent readout [13]. The sandwich format provides high specificity [13].
Protocol Steps:
- Plate Coating:
- Dilute capture antibody in carbonate-bicarbonate buffer (pH 9.4) to 2-10 μg/mL.
- Add 100 μL/well to polystyrene microplate.
- Incubate overnight at 4°C or 2 hours at 37°C.
- Blocking:
- Remove coating solution and wash plate 3 times with PBS-Tween.
- Add 200 μL/well blocking buffer (1% BSA or 5% non-fat dry milk in PBS).
- Incubate 1-2 hours at room temperature.
- Sample and Standard Incubation:
- Prepare standards in sample matrix.
- Add 100 μL/well of standards or samples.
- Incubate 2 hours at room temperature with shaking.
- Detection Antibody Incubation:
- Wash plate 3 times with PBS-Tween.
- Add detection antibody (HRP-conjugated) diluted in blocking buffer.
- Incubate 1-2 hours at room temperature.
- Signal Detection:
- Wash plate 3-5 times with PBS-Tween.
- Add substrate solution (TMB for HRP).
- Incubate 15-30 minutes in dark.
- Stop reaction with stop solution.
- Measure absorbance at appropriate wavelength.
Optimization Strategies for Enhanced Sensitivity:
- Surface Modification: Use protein A/G for oriented antibody immobilization [41], or PEG-grafted copolymers to reduce non-specific binding [41].
- Signal Amplification: Implement tyramide signal amplification or enzyme cycling for enhanced detection [41].
- Alternative Platforms: Consider magnetic beads for improved washing efficiency [41].
Research Reagent Solutions
Table 3: Essential Materials for Advanced Biomarker Analysis
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Capture Molecules | Protein A/G, specific monoclonal/polyclonal antibodies | Target recognition and immobilization; Protein A/G ensures proper antibody orientation [41] |
| Labels and Detection Reagents | Ruthenium tris-bipyridine tags (MSD), HRP, alkaline phosphatase | Signal generation; Ruthenium enables electrochemiluminescence detection in MSD [68] |
| Solid Supports | MSD MULTI-ARRAY plates, polystyrene microplates, magnetic beads | Platform for assay immobilization; MSD plates contain embedded electrodes for signal excitation [67] |
| Separation Media | C18 LC columns, solid-phase extraction cartridges | Sample cleanup and separation; Essential for LC-MS/MS to remove matrix interferents [70] |
| Internal Standards | Isotopically labeled peptides (13C, 15N), isodesmosine-13C3,15N1 | Normalization and precise quantification; Critical for accurate LC-MS/MS analysis [70] |
| Blocking Agents | BSA, casein, non-fat dry milk, synthetic polymers (PEG) | Reduce non-specific binding; PEG-based coatings provide effective nonfouling surfaces [41] |
The evolution of biomarker validation technologies beyond traditional ELISA represents a significant advancement in analytical science. MSD and LC-MS/MS platforms each offer distinct advantages that address specific limitations of conventional ELISA. MSD provides enhanced sensitivity, reduced sample requirements, and efficient multiplexing within the familiar immunoassay framework, making it ideal for targeted biomarker panels in drug development. LC-MS/MS delivers unparalleled specificity, the ability to detect modifications and isoforms, and extensive multiplexing capabilities, positioning it as the gold standard for definitive biomarker identification and validation. Selection between these platforms should be guided by specific research needs, considering factors including required sensitivity, multiplexing scale, target specificity, available sample volume, and budgetary constraints. As regulatory standards continue to evolve toward requiring more comprehensive validation data, embracing these advanced methodologies will be crucial for accelerating biomarker qualification and advancing precision medicine initiatives.
Conclusion
ELISA remains a cornerstone technique for protein detection and quantification, but its reliability is directly dependent on meticulous optimization and validation. By systematically addressing foundational principles, methodological refinements, proactive troubleshooting, and rigorous performance assessment, researchers can significantly enhance assay sensitivity, specificity, and reproducibility. The future of ELISA lies in the adoption of advanced optimization algorithms, such as the Taguchi method, and a clear understanding of its position within the broader analytical landscape, which includes emerging multiplex technologies like MSD. Embracing these comprehensive practices ensures that ELISA will continue to yield high-quality, actionable data, thereby accelerating discoveries in biomedical research and improving the accuracy of clinical diagnostics.
References
- 1. ELISA Evolution | MyBioSource Learning Center [https://www.mybiosource.com/learn/elisa-evolution/]
- 2. The history of ELISA and its enduring role in modern ... [https://www.abcam.com/en-us/stories/articles/the-history-of-elisa]
- 3. Optimizing your ELISA Assays [https://www.bmglabtech.com/en/blog/optimizing-your-elisa-assays/]
- 4. An overview of ELISA: a review and update on best laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11808753/]
- 5. How to Analyze ELISA Data and Calculate Results [https://www.bosterbio.com/blog/post/how-to-analyze-elisa-data-and-calculate-results-step-by-step-guide-with-troubleshooting-tips?srsltid=AfmBOoqPjNo1azn0EsJ7uaIDpQ0bjQKzQjv6e3szK69lGWefoojolWvX]
- 6. Complete ELISA Guide: Get Reliable Results Every Time [https://www.assaygenie.com/complete-elisa-guide-get-reliable-results-every-time-expert-protocols/?srsltid=AfmBOopVyVGsfeFnlE74yIdLh6KEQA1cCZs7IkuV-NYrFEOlZXjylgkU]
- 7. ELISA Development and Optimization [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-development-optimization.html]
- 8. ELISA Guide; Part 3: ELISA Optimization [https://www.jacksonimmuno.com/secondary-antibody-resource/immuno-techniques/elisa-guide-part-3-elisa-optimization/]
- 9. Data analysis for ELISA [https://www.abcam.com/en-us/technical-resources/guides/elisa-guide/calculating-elisa-data]
- 10. The interaction of the antibody molecule with specific antigen [https://www.ncbi.nlm.nih.gov/books/NBK27160/]
- 11. Antigen-antibody interaction [https://en.wikipedia.org/wiki/Antigen-antibody_interaction]
- 12. An ELISA protocol to improve the accuracy and reliability of ... [https://www.sciencedirect.com/science/article/pii/S2215016117300122]
- 13. Overview of ELISA | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 14. ELISA Guide; Part 2: The ELISA Protocol [https://www.jacksonimmuno.com/secondary-antibody-resource/immuno-techniques/elisa-guide-part-2/]
- 15. What is ELISA: Assay and Applications [https://www.moleculardevices.com/applications/enzyme-linked-immunosorbent-assay-elisa]
- 16. Step-by-step guide to ELISA | Mabtech [https://www.mabtech.com/knowledge-hub/step-step-guide-elisa]
- 17. Enzyme Linked Immunosorbent Assay - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK555922/]
- 18. How to Read and Analyze ELISA Data [https://www.rndsystems.com/resources/how-to-analyze-elisa-data]
- 19. What is an ELISA & Types of ELISA Tests [https://www.rndsystems.com/resources/what-is-an-elisa-and-elisa-types]
- 20. Explore ELISA Types: Direct, Indirect, Sandwich, and More [https://www.bosterbio.com/blog/post/elisa-types-indirect-direct-sandwich-competitive-multiplex-elisa?srsltid=AfmBOortnHnNBDHl9PZYvQa2N7WX0FvTCx68CiOLwKFYcdzKJlJx5Vq6]
- 21. ELISA Kits Market Report | Global Forecast From 2025 To ... [https://dataintelo.com/report/global-elisa-kits-market]
- 22. Complete ELISA Guide: Get Reliable Results Every Time [https://www.assaygenie.com/complete-elisa-guide-get-reliable-results-every-time-expert-protocols/?srsltid=AfmBOopKsBij-2wS5h5iUHy3QaEMXQZ0P6PI4ihxjIaEtbZSJiFV8NZO]
- 23. Next-Generation ELISA (ELISA 2.0) Market Research ... [https://www.insightaceanalytic.com/report/next-generation-elisa-market/3241]
- 24. ELISA Buffers and Reagents [https://www.thermofisher.com/us/en/home/life-science/antibodies/immunoassays/elisa-kits/elisa-blocking-buffers-reagents.html]
- 25. Blocking Buffers for Western Blot and ELISA [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-buffers-western-blot-elisa.html]
- 26. Choosing the Right ELISA Blocking Buffer [https://resources.biomol.com/biomol-blog/choosing-the-right-elisa-blocking-buffer]
- 27. ELISA Sample Preparation Guide [https://www.bosterbio.com/protocol-and-troubleshooting/elisa-sample-preparation-guide?srsltid=AfmBOooxv43UTLzn6inwkwM_YwmvB162Y8jv2ff0OoUmJBo9p96amZz-]
- 28. Challenges in the Development and Optimization of Elisa ... [https://www.mybiosource.com/learn/challenges-in-the-development-and-optimization-of-elisa-assay/]
- 29. An ELISA protocol to improve the accuracy and reliability of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5387895/]
- 30. ELISA Development and Optimization [https://www.thermofisher.com/ag/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-development-optimization.html]
- 31. ELISA Optimization [https://www.cytodiagnostics.com/pages/elisa-optimization?srsltid=AfmBOorC6jLZePTywDc7zR4wTrAksEbiXnaANxUDZ7YsSQyPRpmvzNfA]
- 32. Use of the Taguchi Method to Optimize an ... [https://www.mdpi.com/2075-4418/11/7/1179]
- 33. How to Analyze ELISA Data and Calculate Results [https://www.bosterbio.com/blog/post/how-to-analyze-elisa-data-and-calculate-results-step-by-step-guide-with-troubleshooting-tips?srsltid=AfmBOoqzC6nMaqVf_aJykgLU-75ddONWdnLUIUPOv7doLp2Rwixq-jr1]
- 34. Development and Optimization of In-house ELISA for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7601663/]
- 35. Detection and Quantification of Cytokines and Other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5659280/]
- 36. Cytokine ELISA Protocol [https://www.bdbiosciences.com/en-us/resources/protocols/cytokine-elisa]
- 37. ELISA Troubleshooting Guide [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-troubleshooting-guide.html]
- 38. ELISA: The Complete Guide [https://www.antibodies.com/applications/elisa]
- 39. How to deal with high background in ELISA [https://www.abcam.com/en-us/technical-resources/troubleshooting/high-background-elisa]
- 40. What Causes High Background in ELISA Tests? [https://shop.surmodics.com/elisa-test-results-high-background-causes]
- 41. Enhancing ELISA Sensitivity: From Surface Engineering to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293621/]
- 42. Causes Of High Background In ELISA Tests and How to ... [https://www.caltagmedsystems.co.uk/information/causes-of-high-background-in-elisa-tests-and-how-to-solve-them/]
- 43. ELISA Sensitivity and Specificity [https://www.mybiosource.com/learn/elisa-sensitivity-and-specificity/]
- 44. ELISA Data Analysis | Thermo Fisher Scientific - ES [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-data-analysis.html]
- 45. When to use Triplicates, Duplicates, or Single ... [https://www.enzo.com/note/when-to-use-triplicates-duplicates-or-single-measurements-in-elisa-and-why/]
- 46. Common ELISA Problems and Solutions [https://www.myassays.com/common-elisa-problems-and-solutions.html]
- 47. Development of an enzyme-linked immunosorbent assay ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1542898/full]
- 48. How to Troubleshoot High Background Noise in ELISA [https://www.linkedin.com/advice/0/how-do-you-troubleshoot-high-background-noise]
- 49. How To Optimize Your ELISA Experiments | Proteintech Group [https://www.ptglab.com/news/blog/how-to-optimize-your-elisa-experiments/]
- 50. Improved immunoassay sensitivity and specificity using ... [https://www.nature.com/articles/s41467-022-32796-x]
- 51. the Optimizing (Enzyme-Linked...) | Cell Signaling Technology ELISA [https://www.cellsignal.com/applications/elisa/optimizing-elisa-test]
- 52. Optimizing the ELISA (Enzyme-Linked Immunosorbent ... [https://www.cellsignal.com/applications/elisa/optimizing-elisa-test?srsltid=AfmBOoph7ZoPMQBOiI9Y1YajvQccu0lHWxlRG0ZyFyBU2bvFxd49sajI]
- 53. Impact of high temperatures on enzyme-linked immunoassay ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0320366]
- 54. 101 ELISA Troubleshooting Tips for Research in 2024 [https://www.assaygenie.com/101-elisa-troubleshooting-tips?srsltid=AfmBOoojtaejr3YjEuN6bq_eTltZiDYDAccBIdJMC1kw0AshJ6MC-Mdx]
- 55. Step-by-Step ELISA Protocol: A Comprehensive Guide [http://www.genfollower.com/step-by-step-elisa-protocol/]
- 56. Validation of Elisa protocol as per FDA Regulation for Drug ... [https://www.mybiosource.com/learn/validation-of-elisa-protocol-as-per-fda-regulation-for-drug-developmen/]
- 57. ELISA Assay Development and Validation: A Complete Guide [https://protocolsandsolutions.com/blogs/news/elisa-assay-development-and-validation-a-complete-guide-%E2%9C%85]
- 58. Understanding ELISA Validation Parameters: Sensitivity ... [https://synapse.patsnap.com/article/understanding-elisa-validation-parameters-sensitivity-specificity-and-beyond]
- 59. Spike and Recovery and Linearity of Dilution Assessment [https://www.thermofisher.com/ng/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/spike-recovery-linearity-assessment.html]
- 60. Establishing Dilution Linearity for Your Samples in an ELISA [https://www.cygnustechnologies.com/establishing-dilution-linearity-for-your-samples-in-an-elisa?srsltid=AfmBOooaKYyvOLxPOMapEyFmlzD8kqTrTZ5gwD9F7kP36AQF538_vmGi]
- 61. Spike and Recovery: How to Generate Reliable Data [https://www.cygnustechnologies.com/generating-spike-recovery-analysis-data?srsltid=AfmBOoou-9qicnS1E_D4iUf9YgYOkb-E97TxPfe2tvuS0IeArb1wmpsw]
- 62. How To Perfect Your ELISA Standard Curve [https://www.bosterbio.com/blog/post/how-to-perfect-your-elisa-standards?srsltid=AfmBOoqoveyn85d-wDUdZ0yElwiULvX9F0-nKp1Zxswebrz2RRZfkOX0]
- 63. Assay Optimization 101: Enhancing Sensitivity, Specificity, ... [https://dispendix.com/blog/assay-optimization-101-enhancing-sensitivity-specificity-and-reproducibility]
- 64. How to Obtain Reproducible ELISA Assay Results - Biomedica [https://www.bmgrp.com/how-to-obtain-reproducible-elisa-assay-results/]
- 65. Which Controls to Use in ELISA Assays? [https://www.enzo.com/note/which-controls-to-use-in-elisa-assays/]
- 66. Beyond ELISA: the future of biomarker validation [https://www.drugtargetreview.com/article/154316/beyond-elisa-the-future-of-biomarker-validation/]
- 67. ELISA Comparison [https://www.mesoscale.com/en/technical_resources/our_technology/our_immunoassays/elisa_comparison]
- 68. Protein Quantitation: Finding the Right Assay for Your Needs [https://emerypharma.com/blog/protein-quantitation-finding-the-right-assay-for-your-needs/]
- 69. The advantages and the limitations of LC-MS/MS and Elisa ... [https://www.biotrial.com/bioanalysis-lc-ms-ms-vs-elisa/]
- 70. Correlation between LC-MS/MS and ELISA methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536777/]
- 71. Mass Spectometry: The Future of Impurity Analysis [https://www.bioprocessintl.com/sponsored-content/beyond-elisa-is-mass-spectrometry-the-future-of-impurity-analysis-]
- 72. Multiplex assays for biomarker research and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248403/]