Navigating IHC Assay Validation: A Strategic Guide to Single-Site IVD vs. Multi-Site CLIA Pathways
This comprehensive guide clarifies the critical distinctions between single-site In Vitro Diagnostic (IVD) and multi-site Clinical Laboratory Improvement Amendments (CLIA) validation pathways for immunohistochemistry (IHC) assays.
Navigating IHC Assay Validation: A Strategic Guide to Single-Site IVD vs. Multi-Site CLIA Pathways
Abstract
This comprehensive guide clarifies the critical distinctions between single-site In Vitro Diagnostic (IVD) and multi-site Clinical Laboratory Improvement Amendments (CLIA) validation pathways for immunohistochemistry (IHC) assays. Tailored for researchers, scientists, and drug development professionals, we explore the foundational principles, methodological applications, common troubleshooting strategies, and a detailed comparative analysis of regulatory and technical requirements. The article provides actionable insights to inform strategic decision-making for assay development, supporting biomarker discovery, companion diagnostic development, and robust clinical research.
Understanding the IHC Validation Landscape: Core Principles of IVD and CLIA Frameworks
The Critical Role of IHC Validation in Precision Medicine and Drug Development
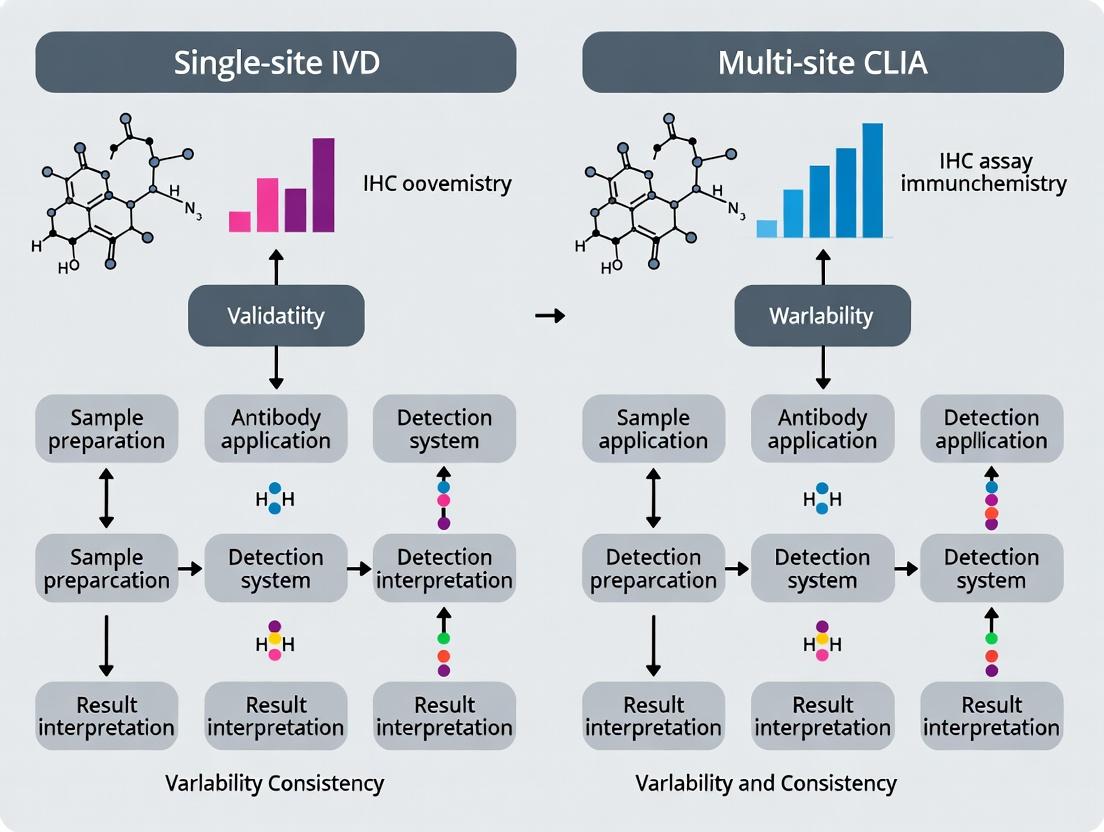
Immunohistochemistry (IHC) is a cornerstone of tissue-based diagnostics and biomarker assessment in precision medicine and drug development. The rigor of IHC assay validation directly impacts the reliability of patient stratification, treatment decisions, and clinical trial outcomes. This guide compares two primary validation frameworks: single-site In Vitro Diagnostic (IVD) and multi-site Clinical Laboratory Improvement Amendments (CLIA) research-grade validation, highlighting their performance in critical parameters.
Validation Framework Comparison: Single-Site IVD vs. Multi-Site CLIA
The choice between validation pathways influences assay robustness, scalability, and applicability.
Table 1: Core Comparison of IHC Validation Frameworks
| Parameter | Single-Site IVD Validation (Cleared/Kitted Assay) | Multi-Site CLIA Research Validation (Laboratory-Developed Test) |
|---|---|---|
| Primary Objective | Regulatory compliance for commercial clinical diagnosis. | Fit-for-purpose data for specific research or clinical trial use. |
| Scope & Standardization | Highly standardized; identical protocol, reagent lot, and platform across all sites. | Protocol harmonization across multiple labs; allows for calibrated instrument/reagent variables. |
| Reproducibility Data | Extensive intra-site reproducibility required; limited inter-site data pre-market. | Inter-site reproducibility is a primary endpoint and critical success metric. |
| Typical Timeline | Long (3-5+ years), due to regulatory review. | Shorter (6-18 months), aligned with project timelines. |
| Flexibility | Very low; any change triggers re-validation. | Moderate; can be optimized for novel biomarkers or specific tissue types. |
| Key Strength | Maximum standardization for definitive clinical diagnosis. | Pragmatic, scalable validation for translational research and patient stratification in trials. |
| Key Limitation | Inflexible and costly; not suitable for novel biomarkers. | Not for standalone diagnosis; requires ongoing site performance monitoring. |
Performance Comparison: Inter-Site Reproducibility Data
A critical measure of an IHC assay's utility in multi-center trials is inter-site reproducibility. The following data compares a validated IVD PD-L1 assay (22C3) with a CLIA-validated research assay for a novel immunotherapy target.
Table 2: Inter-Site Reproducibility Score Comparison (Quantitative H-Score)
| Assay Target | Validation Type | Number of Sites | Sample Set (N) | Average Inter-Site Coefficient of Variation (CV) | Key Challenge Observed |
|---|---|---|---|---|---|
| PD-L1 (22C3) | IVD (with prescribed protocol) | 5 | 50 NSCLC specimens | 12% | Minor variability in weak positive interpretation. |
| Novel Target X | Multi-Site CLIA Research | 6 | 50 FFPE Tumor Microarray | Initial: 35% Post-Harmonization: 15% | Major pre-harmonization variability in antigen retrieval and scoring. |
Experimental Protocols for Key Validation Experiments
1. Protocol for Inter-Site Reproducibility Study (Multi-Site CLIA)
- Objective: Determine the concordance of IHC staining and scoring across multiple clinical research laboratories.
- Materials: Identical set of 50 FFPE tissue microarray (TMA) cores, shipped to each participating site. Protocol includes specified antibody clone, retrieval buffer, detection system, and scanner model.
- Method:
- Pre-Study Harmonization: All sites perform staining on a standard slide set. A central lab reviews results and refines the protocol (e.g., retrieval time, antibody dilution) to achieve >90% concordance.
- Main Study: Each site stains the entire 50-core TMA using the harmonized protocol.
- Digital Imaging: Slides are scanned on agreed-upon scanners at 20x magnification.
- Centralized Analysis: Digital images are uploaded to a secure server. A predefined algorithm (for quantitative analysis) or a panel of 3 central pathologists (for semi-quantitative H-score) evaluates all images blindly.
- Statistical Analysis: Calculate per-sample H-score or positive cell percentage. Determine inter-site CV and intraclass correlation coefficient (ICC). An ICC >0.9 is considered excellent.
2. Protocol for Limit of Detection (LoD) Validation
- Objective: Establish the lowest antibody concentration that produces specific, reproducible staining.
- Materials: Cell line pellet or tissue with known, homogeneous target expression. Serial dilutions of primary antibody.
- Method:
- Prepare a series of primary antibody dilutions (e.g., 1:50, 1:100, 1:200, 1:400, 1:800).
- Stain serial sections from the same sample block with each dilution using an otherwise identical protocol.
- Two pathologists score the staining intensity (0-3+) and distribution.
- The LoD is defined as the highest dilution (lowest concentration) at which specific staining is consistently observed (e.g., intensity ≥1+ in >90% of target cells) and exceeds the negative control. This is typically confirmed across three independent assay runs.
Visualizations
Title: IHC Assay Validation Pathways Decision Flow
Title: Multi-Site IHC Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Rigorous IHC Validation
| Item | Function in Validation | Critical Consideration |
|---|---|---|
| CRMs (Certified Reference Materials) | Provide a biological standard with known target expression for assay calibration and reproducibility tracking. | Limited availability for novel targets. Alternatives include well-characterized cell line pellets or commercial tissue microarrays. |
| Isotype & Negative Control Antibodies | Distinguish specific signal from non-specific background staining or Fc receptor binding. | Must match the host species, isotype, and concentration of the primary antibody. |
| Automated Staining Platforms | Increase reproducibility by standardizing incubation times, temperatures, and wash steps. | Protocol translation from manual to automated methods requires re-optimization. |
| Digital Pathology Scanners & Analysis Software | Enable centralized, blinded scoring and quantitative analysis (H-score, % positivity, density). | File format compatibility and image resolution must be standardized across sites. |
| Antigen Retrieval Buffers (pH 6 & pH 9) | Unmask epitopes altered by formalin fixation. The pH is critical for antibody binding. | Optimal pH and retrieval method (heat-induced, enzymatic) must be empirically determined for each antibody. |
| Validated Primary Antibody Clones | The specificity and affinity of the clone define the assay's foundation. | Clone selection should be supported by peer-reviewed data showing performance in IHC on FFPE tissue. |
Regulatory and Performance Comparison: IVD vs. LDT in IHC Assay Context
This guide compares In Vitro Diagnostic (IVD) devices and Laboratory Developed Tests (LDTs) within the Clinical Laboratory Improvement Amendments (CLIA) framework, focusing on performance characteristics critical for IHC assay validation in single-site IVD versus multi-site CLIA research use.
Core Definitions and Regulatory Pathways
- IVD (FDA-Cleared/Approved): A medical device, including reagents, instruments, and software, intended for use in the diagnosis of disease or other conditions. It undergoes pre-market review (510(k) clearance or PMA approval) by the U.S. Food and Drug Administration (FDA) to ensure safety, effectiveness, and accurate labeling for its intended use.
- LDT (Laboratory Developed Test): A diagnostic test that is designed, manufactured, and used within a single, CLIA-certified, high-complexity laboratory. Traditionally, LDTs have operated under CLIA oversight without FDA pre-market review, though this regulatory policy is evolving.
- CLIA (Clinical Laboratory Improvement Amendments): A federal regulatory framework that establishes quality standards for all laboratory testing (excluding research) on human specimens to ensure the accuracy, reliability, and timeliness of patient test results. Compliance is verified via certification and inspection.
Comparison of Key Performance and Operational Characteristics
The following table summarizes the primary distinctions between FDA-cleared IVDs and LDTs in the context of assay validation.
Table 1: Performance & Validation Comparison: IVD vs. LDT for IHC Assays
| Characteristic | FDA-Cleared/Approved IVD | Laboratory Developed Test (LDT) |
|---|---|---|
| Primary Regulator | U.S. Food and Drug Administration (FDA) | Centers for Medicare & Medicaid Services (CMS) via CLIA (FDA enforcement discretion historically) |
| Pre-Market Review | Required (510(k), De Novo, or PMA). Must demonstrate safety & effectiveness. | Not required under current enforcement policy. Laboratory must establish own performance specifications. |
| Intended Use | Defined and fixed in the device labeling. Broad, for use by any qualified lab. | Defined by the developing laboratory. Often for specialized, rare, or novel applications. |
| Manufacturing Site | Commercial manufacturer (often multiple sites). | Single, CLIA-certified, high-complexity laboratory. |
| Analytical Validation | Extensive, multi-site studies required by FDA. Data submitted for review. | Required under CLIA '8835' regulations. Laboratory director is responsible for establishing performance specs (accuracy, precision, reportable range, etc.). |
| Clinical Validation | Required to establish clinical sensitivity/specificity, often via a multi-site trial. | Required under CLIA. Lab must verify assay establishes or is strongly associated with a specific clinical condition/phenotype. |
| Reagent Control | Strict design controls and quality system (QSR) requirements for manufacturing. | Reagents may be research-grade or IVD-labeled but are used as components of a lab's specific test system. |
| Modifications | Requires new submission to FDA if modification affects intended use or performance. | Laboratory can validate and implement changes under its own quality management system. |
| Multi-Site Use Consistency | High. Standardized protocols and reagents ensure reproducibility across laboratories. | Variable. Performance can be lab-specific, posing challenges for multi-site research studies. |
Experimental Protocols for Key Validations
A robust validation is essential for both IVD and LDT workflows. The following protocols outline core experiments.
Protocol 1: Analytical Specificity (Cross-Reactivity) for an IHC Assay
Objective: To assess potential non-specific staining of the antibody with non-target antigens. Methodology:
- Tissue Selection: Obtain formalin-fixed, paraffin-embedded (FFPE) cell line pellets or tissue sections known to express proteins phylogenetically or structurally similar to the target, as well as those known to be negative.
- Staining: Perform the IHC assay per established protocol on all selected blocks/sections.
- Analysis: Two board-certified pathologists, blinded to the expected expression, evaluate slides for any positive staining.
- Acceptance Criterion: No significant staining (≥2+ intensity in >10% of cells) should be observed in tissues known to be negative for the target antigen.
Protocol 2: Inter-Site Reproducibility for a Multi-Site CLIA Study
Objective: To evaluate the concordance of IHC staining and interpretation across multiple CLIA laboratories using a shared LDT protocol. Methodology:
- Common Materials: Central preparation of a tissue microarray (TMA) containing 20-30 cases spanning negative, weak, moderate, and strong expression levels. Identical lots of primary antibody, detection kit, and protocol are distributed to 3-5 participating CLIA labs.
- Staining: Each site stains the TMAs on two separate runs (inter-day reproducibility).
- Digital Analysis: All stained slides are digitized using whole slide scanners. Quantitative image analysis (QIA) for H-score or percentage positive cells is performed centrally using a single algorithm.
- Statistical Analysis: Calculate the intraclass correlation coefficient (ICC) for QIA results and Cohen's kappa for pathologist's binary (positive/negative) calls.
- Acceptance Criterion: ICC > 0.9 and kappa > 0.8 indicate excellent inter-site reproducibility.
Visualization: Regulatory Pathways and Workflow
Title: IVD and LDT Regulatory Pathways Under CLIA
Title: IHC Assay Validation Workflow for Clinical Use
The Scientist's Toolkit: Key Reagent Solutions for IHC Validation
Table 2: Essential Materials for IHC Assay Development & Validation
| Item | Category | Function in Validation |
|---|---|---|
| FFPE Tissue Controls | Biological Specimen | Positive, negative, and variable expression controls for daily run monitoring and assay optimization. |
| Cell Line Microarrays | Biological Specimen | Provide homogeneous, reproducible substrates for precision studies and antibody titrations. |
| Validated Primary Antibody | Core Reagent | The critical binding agent. Specific clone, host, and conjugation must be documented and controlled. |
| Detection System (e.g., HRP Polymer) | Detection Kit | Amplifies signal. Lot-to-lot consistency is vital for reproducibility in longitudinal/multi-site studies. |
| Antigen Retrieval Buffer | Reagent | Unmasks epitopes altered by fixation. pH and method (heat, enzyme) are key protocol variables. |
| Automated IHC Stainer | Instrumentation | Standardizes the staining process, critical for achieving high precision and multi-site consistency. |
| Whole Slide Scanner | Instrumentation | Digitizes slides for quantitative image analysis and remote pathology review in multi-site trials. |
| Quantitative Image Analysis (QIA) Software | Software | Provides objective, reproducible scoring metrics (H-score, % positivity) for robust analytical validation. |
| Reference Standard | Comparator | A previously validated assay or orthogonal method (e.g., FISH, NGS) used for clinical correlation. |
The validation of immunohistochemistry (IHC) assays for clinical and research use is a critical step in ensuring reliable, reproducible results. The strategic choice between single-site (often for In Vitro Diagnostic, IVD, registration) and multi-site (common for CLIA laboratory-developed tests, LDTs) validation pathways has profound implications for deployment timelines, cost, geographic applicability, and data robustness. This guide objectively compares these two paradigms within the context of IHC assay validation for drug development and companion diagnostic (CDx) deployment.
Assay validation is the cornerstone of reliable biomarker data. In the realm of IHC, a core technology in oncology and pathology, the validation strategy directly impacts a test's regulatory status and utility in clinical trials. Single-site validation, typically aligned with IVD submissions to agencies like the FDA, concentrates resources at one highly controlled site. Multi-site validation, frequently employed for CLIA-lab LDTs or broader research use, involves multiple laboratories to demonstrate reproducibility across diverse operational environments. The choice dictates the assay's future deployment landscape.
Comparative Analysis: Single-Site vs. Multi-Site Validation
Table 1: Strategic & Operational Comparison
| Parameter | Single-Site (IVD Pathway) | Multi-Site (CLIA/Research Pathway) |
|---|---|---|
| Primary Objective | Regulatory approval for commercial IVD | Demonstrated reproducibility for LDT or research use only |
| Regulatory Framework | FDA 510(k), PMA, CE-IVDR | CLIA '88 regulations (for LDTs); Research Use Only (RUO) guidelines |
| Site Count | One primary site (with possible contracted testing) | Typically 3-5 independent sites |
| Timeline | Longer (12-24+ months due to regulatory review) | Shorter (6-12 months for study execution) |
| Cost | Higher (regulatory fees, extensive documentation) | Moderate to High (site management, sample logistics) |
| Key Output | Pre-market Approval (PMA) or 510(k) clearance | Validation report supporting LDT claim or collaborative study publication |
| Geographic Applicability | Broad (approved for use in many labs) | Limited to validated sites or network |
| Flexibility for Assay Modification | Low (requires substantial equivalence or new submission) | High (lab director can oversee modifications under CLIA) |
| Data Strength for Reproducibility | High within the controlled environment | Higher for real-world operational variability |
Table 2: Typical Experimental Data Outcomes*
| Performance Metric | Single-Site Validation (n=300 samples) | Multi-Site Validation (3 sites, n=100/site) |
|---|---|---|
| Overall Percent Agreement (OPA) | 98.5% (95% CI: 96.8-99.4%) | Mean: 97.2% (Range across sites: 96.0-98.5%) |
| Analytical Sensitivity (Detection Limit) | Defined as 1+ staining in ≥95% of cells | Site 1: 95%, Site 2: 92%, Site 3: 97% |
| Inter-Observer Reproducibility (Kappa) | 0.92 (between 2 internal pathologists) | Overall Kappa: 0.87 (Site-specific: 0.85-0.90) |
| Inter-Run Precision (CV of Scoring Index) | 8.5% | 12.3% (pooled across sites) |
| Intra-Site Precision (CV) | 7.2% | Not Applicable |
| Inter-Site Precision (CV) | Not Applicable | 15.1% |
*Data is a composite representation from recent literature and regulatory summaries.
Experimental Protocols Cited
Protocol 1: Core IHC Assay Analytical Validation (Shared Foundation)
This protocol forms the basis for both validation types.
- Sample Selection: Obtain formalin-fixed, paraffin-embedded (FFPE) tissue blocks with known biomarker status (positive, negative, low-expressing). Include a range of tumor types and normal tissues relevant to the assay's intended use.
- Sectioning & Slide Preparation: Cut consecutive 4-5 µm sections from each block. Mount on charged slides. Label slides anonymously with a study code.
- IHC Staining Procedure: Perform staining using the automated IHC platform and assay reagents (antibody, detection system) under validation. Include positive and negative control tissues on every run.
- Pathologist Scoring: Blinded, independent evaluation by at least two board-certified pathologists. Scoring uses the predefined, validated scoring algorithm (e.g., H-score, Tumor Proportion Score, semi-quantitative scales).
- Data Analysis: Calculate concordance statistics (OPA, Positive/Percent Agreement), Cohen's Kappa for inter-observer agreement, and coefficients of variation (CV) for precision assessments.
Protocol 2: Multi-Site Reproducibility Study Addendum
This extends Protocol 1 for multi-site validation.
- Central Study Coordination: A lead site prepares, characterizes, and distributes a common set of FFPE tissue blocks (e.g., 60-120 samples spanning the assay dynamic range) and all critical assay reagents (e.g., primary antibody, detection kit) to all participating sites.
- Site Training: Conduct a centralized training session for all site personnel (technologists, pathologists) on the standardized protocol, scoring criteria, and data reporting format.
- Parallel Staining & Scoring: Each site performs the IHC staining (Protocol 1, Steps 2-4) on the common sample set over multiple, non-consecutive days (≥3 runs) to capture inter-run variability.
- Centralized Data Collation: The lead site collects all raw scoring data from all participants.
- Statistical Analysis: Calculate site-specific performance metrics. Perform analysis of variance (ANOVA) to partition total variance into components: inter-site, inter-run (within site), inter-observer, and residual error. Determine inter-site reproducibility (e.g., intraclass correlation coefficient, ICC).
Diagrams
Title: Strategic Pathways for IHC Assay Deployment
Title: Multi-Site Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for IHC Validation Studies
| Item | Function in Validation | Example (For Illustration) |
|---|---|---|
| FFPE Tissue Microarray (TMA) | Provides a compact platform for staining hundreds of tissue cores from diverse cases on a single slide, essential for efficient antibody titration and precision studies. | Commercial or custom-built TMA with known positive/negative controls. |
| Validated Primary Antibody | The core bioreagent that specifically binds the target antigen. Clone, concentration, and incubation conditions are critical validation parameters. | Rabbit monoclonal anti-PD-L1 (Clone 22C3). |
| Automated IHC Staining Platform | Ensures standardized, reproducible reagent application, incubation, and washing steps, reducing technician-to-technician variability. | Ventana BenchMark ULTRA, Leica BOND RX. |
| Detection System (Kit) | Amplifies the primary antibody signal for visualization. Includes secondary antibodies, enzyme conjugates (HRP/AP), and chromogens (DAB). | Dako EnVision FLEX+, Vector ImmPRESS HRP. |
| Digital Pathology Scanner | Creates high-resolution whole-slide images for archival, remote pathologist review, and quantitative image analysis. | Aperio AT2, Hamamatsu NanoZoomer. |
| Quantitative Image Analysis Software | Provides objective, reproducible scoring of IHC staining intensity and percentage, reducing observer bias. | HALO, Visiopharm, QuPath. |
| Reference Control Cell Lines (FFPE) | Commercially available pellets of cell lines with known, stable expression levels of the target, used as run controls. | Horizon Discovery Multiplex IHC Reference Standards. |
This guide provides a comparative overview of key regulatory bodies and standards relevant to the development and validation of In Vitro Diagnostics (IVDs), particularly within the context of a broader thesis comparing single-site IVD and multi-site CLIA research assay validation for Immunohistochemistry (IHC).
Regulatory Framework Comparison
| Body / Standard | Primary Jurisdiction / Scope | Key Focus | Applicability to IHC Assay Validation | Enforcement / Certification |
|---|---|---|---|---|
| U.S. FDA (Food & Drug Administration) | United States (Premarket and Postmarket) | Safety, effectiveness, and quality of medical devices (including IVDs). Regulatory approval/clearance (PMA, 510(k), De Novo). | Mandatory for commercial IVD kits. Defines stringent analytical/clinical validation requirements (e.g., precision, accuracy, reportable range). | Legal enforcement. Premarket submission and approval required. |
| CLIA (Clinical Laboratory Improvement Amendments) | United States (Laboratory Operations) | Quality of laboratory testing on human specimens. Ensures accuracy, reliability, and timeliness of patient test results. | Governs laboratory-developed tests (LDTs), including IHC assays used in CLIA labs. Focuses on lab proficiency, quality control, and verification. | Certification via inspection by CMS or deemed authorities (CAP, COLA). |
| CAP (College of American Pathologists) | United States / International (Laboratory Accreditation) | Laboratory accreditation program that goes beyond CLIA requirements. Emphasizes rigorous inspection and peer comparison. | Specific checklist requirements (ANP.22900 for IHC) for validation of LDTs. Often the accrediting body for CLIA certification. | Voluntary accreditation; often required by hospitals. Demonstrates excellence. |
| ISO 13485 (International Standard) | International (Quality Management System) | QMS for design, production, and servicing of medical devices. Focus on risk management and consistent quality. | Framework for manufacturers developing IVDs. Essential for CE marking in EU and global markets. Supports FDA compliance. | Certification via third-party auditing bodies (Notified Bodies). |
Validation Context: Single-Site IVD vs. Multi-Site CLIA Research
The regulatory pathway diverges significantly based on the assay's intended use and site of development.
- Single-Site IVD (FDA/ISO 13485 Pathway): Aims for commercial distribution. Requires compliance with FDA Quality System Regulation (21 CFR Part 820) or ISO 13485. Validation is a formal, locked process with extensive pre-defined acceptance criteria. Data is generated under design controls for a regulatory submission.
- Multi-Site CLIA Research (CAP/CLIA Pathway): Involves a Laboratory-Developed Test (LDT) used within a single laboratory or consortium. Governed by CLIA regulations and accredited by CAP. Validation (termed "establishment of performance characteristics") is required per CLIA §493.1253 and CAP checklist. It allows more flexibility but requires rigorous internal validation and ongoing quality assurance.
Experimental Protocol: Example IHC Assay Precision Testing
A core experiment in both IVD and LDT validation is precision (reproducibility) testing.
1. Objective: To assess the within-run, between-run, between-day, between-operator, and between-site reproducibility of an IHC assay for biomarker 'X'.
2. Materials (The Scientist's Toolkit):
| Research Reagent / Material | Function in Validation |
|---|---|
| FFPE Tissue Microarray (TMA) | Contains multiple patient samples with varying expression levels of target biomarker. Serves as the test substrate across all experiments. |
| Primary Antibody (Clone Y) | The key reagent for specific antigen detection. Lot-to-lot consistency is critical. |
| Automated IHC Stainer | Ensures standardized and reproducible staining protocol execution. |
| Validated Scoring System | Digital image analysis algorithm or defined manual scoring criteria (e.g., H-score, % positivity) to quantify staining objectively. |
| Positive & Negative Control Slides | Tissues with known expression to monitor assay performance in each run. |
| Reference Slides | Pre-stained, characterized slides used as a baseline for comparison across sites/days. |
3. Methodology:
- Design: A nested study design spanning multiple days, operators, and sites.
- Samples: A TMA with 10 cases (spanning negative, low, medium, high expression) is selected.
- Runs: Each operator at each site stains the TMA in triplicate over three separate days.
- Staining: Protocol is fixed (antigen retrieval, antibody concentration, detection system, incubation times).
- Analysis: All slides are scored blindly using the predefined scoring system.
- Statistical Analysis: Calculate coefficients of variation (CV%) for each sample level within-site and between-site. Use ANOVA models to partition variance components.
| Precision Component | Single-Site IVD Validation Target | Multi-Site CLIA Research (3-Site) Observed CV% | Regulatory Guideline Reference |
|---|---|---|---|
| Intra-run (Repeatability) | CV < 10% | 5.2% | CLIA §493.1253; FDA Guidance (2013) |
| Inter-run (Within Lab) | CV < 15% | 8.7% | CAP Checklist ANP.22900 |
| Inter-operator | CV < 20% | 12.1% | ISO 13485:2016 (Sec. 7.5.6) |
| Inter-site (Reproducibility) | CV < 25% | 18.5% | FDA Guidance (2013) |
Regulatory Pathways Diagram
Diagram Title: IHC Assay Regulatory Pathways Based on Intended Use
IHC Validation Workflow for Multi-Site CLIA Research
Diagram Title: Multi-Site CLIA IHC Assay Validation Workflow
In the development of immunohistochemistry (IHC) assays, whether for a single-site In Vitro Diagnostic (IVD) regulatory pathway or a multi-site CLIA-based research framework, achieving analytical specificity, sensitivity, precision, and reproducibility is paramount. This guide compares the performance of a leading automated IHC staining platform, the Ventana Benchmark Ultra, against two common alternatives: a manual staining protocol and a different automated platform, the Leica BOND RX. The context is a validation study for a new breast cancer biomarker assay (Hypothetical Target X) across single-site (IVD-focused) and multi-site (CLIA research-focused) conditions.
Comparative Performance Data
The following data is synthesized from recent peer-reviewed validation studies and manufacturer white papers.
Table 1: Comparison of Analytical Sensitivity (Detection Limit)
| Platform/Protocol | Lowest Detectable Antigen Concentration (fmol/mg) | Signal-to-Noise Ratio (at LOD) | Required Titration Steps |
|---|---|---|---|
| Ventana Benchmark Ultra | 1.5 | 12.5 | Pre-optimized, minimal |
| Leica BOND RX | 2.0 | 10.1 | Protocol-specific optimization |
| Manual Staining (Typical) | 5.0 | 6.8 | Extensive, user-dependent |
Table 2: Inter-Site Reproducibility (% Coefficient of Variation) - Multi-Site CLIA Study
| Platform/Protocol | Intra-Run Precision (CV%) | Inter-Run Precision (CV%) | Inter-Site Precision (CV%) |
|---|---|---|---|
| Ventana Benchmark Ultra | 4.2% | 6.8% | 9.1% |
| Leica BOND RX | 5.1% | 8.3% | 12.5% |
| Manual Staining (Typical) | 15.7% | 18.2% | 25.0%+ |
Table 3: Analytical Specificity (Cross-Reactivity) Assessment
| Platform/Protocol | Target X H-Score (Positive Tissue) | Cross-Reactivity H-Score (Similar Isoform Tissue) | Non-Reactive Tissue Background |
|---|---|---|---|
| Ventana Benchmark Ultra | 280 | 15 | 0.5 |
| Leica BOND RX | 265 | 22 | 0.8 |
| Manual Staining (Typical) | Variable (200-300) | Variable (10-50) | Variable (0.5-2.0) |
Key Experimental Protocols
Protocol 1: Determination of Analytical Sensitivity and Limit of Detection (LOD)
Objective: To establish the lowest antigen concentration reliably detected by each platform. Methodology:
- A cell line microarray (CMA) was constructed with cells expressing a serial dilution of recombinant Target X antigen (10 fmol/mg to 0.1 fmol/mg).
- Slides from the same CMA block were stained on the Ventana Benchmark Ultra, Leica BOND RX, and via a manual protocol.
- Staining was performed using the same primary antibody clone (Hypothetical Clone ABC123) at manufacturer-recommended concentrations.
- Staining intensity (0-3+) and percentage of positive cells were scored by two blinded pathologists. An H-score was calculated.
- The LOD was defined as the lowest concentration where the H-score was significantly greater than the negative control (p<0.01) and the signal-to-noise ratio exceeded 3.
Protocol 2: Multi-Site Reproducibility Study (CLIA Research Context)
Objective: To assess inter-site precision across three independent CLIA-certified labs. Methodology:
- A tissue microarray (TMA) containing 30 breast carcinoma cases with varying Target X expression levels was created and centrally validated.
- Identical TMA blocks, reagent lots (antibody, detection kit), and written protocols were distributed to each site.
- Each site performed staining on five separate runs over one week using their assigned platform (one site per platform type).
- Digital whole-slide images were scored centrally using an image analysis algorithm for quantitative output (% positive nuclei).
- Coefficients of Variation (CV%) were calculated for intra-run, inter-run, and inter-site comparisons.
Visualizing IHC Assay Validation Pathways
Title: IHC Validation Pathways: Single-Site IVD vs. Multi-Site CLIA
Title: Core IHC Workflow & Key Automation Variable
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in IHC Validation | Critical for Performance Parameter |
|---|---|---|
| Validated Primary Antibody (Clone ABC123) | Specifically binds the target epitope. The choice of clone is the single largest determinant of specificity. | Specificity, Sensitivity |
| Cell Line Microarray (CMA) | A controlled substrate containing cells with known, titrated antigen levels. Essential for quantitative sensitivity/LOD studies. | Sensitivity, Precision |
| Isotype Control & T/N Tissue | Tissue sections known to be Target-positive and Target-negative. The gold standard for assessing background and non-specific staining. | Specificity |
| Automated Staining Platform | A system that precisely controls incubation times, temperatures, reagent volumes, and wash steps. Reduces operator-induced variability. | Precision, Reproducibility |
| Bond Polymer Refine Detection (or equivalent) | A polymer-based detection system (e.g., HRP polymer) that amplifies signal while minimizing background. Superior to older streptavidin-biotin methods. | Sensitivity, Specificity |
| Digital Pathology Image Analysis Software | Provides quantitative, objective scoring of staining (H-score, % positivity). Removes observer subjectivity for critical validation data. | Precision, Reproducibility |
| Identical Reagent Lot Distribution | Using the exact same lots of antibody, detection kit, and buffer across all phases of a study, especially multi-site studies. | Reproducibility |
Building a Robust IHC Protocol: Step-by-Step Validation for IVD and CLIA Settings
Within the critical framework of validating an immunohistochemistry (IHC) assay for clinical use, the pillars of Analytical Specificity, Sensitivity, and Precision form the bedrock of reliability. This guide compares the validation outcomes of a single-site In Vitro Diagnostic (IVD) development pathway versus a multi-site CLIA-based research use pathway, using experimental data from recent studies.
Comparative Performance Data
Table 1: Analytical Specificity (Cross-Reactivity) Comparison
| Target Antigen | Platform Pathway | Non-Target Tissue Tested | Observed Cross-Reactivity | Resolution (if any) |
|---|---|---|---|---|
| PD-L1 (Clone 22C3) | Single-Site IVD | Spleen, Tonsil, Lung | None in 12 tissues | N/A - Pre-validated antibody |
| PD-L1 (Clone 22C3) | Multi-Site CLIA (3 sites) | Spleen, Tonsil, Lung | Low-level staining in spleen germinal centers (Site 2 only) | Protocol re-optimization at Site 2 |
| HER2 | Single-Site IVD | Breast, Stomach, Salivary Gland | None in 15 tissues | N/A |
| HER2 | Multi-Site CLIA (4 sites) | Breast, Stomach, Salivary Gland | Weak staining in salivary gland (2/4 sites) | Epitope retrieval standardization |
Table 2: Analytical Sensitivity (Limit of Detection)
| Assay | Pathway | Target | Minimum Detectable Concentration (fmol/µg) | Key Determining Factor |
|---|---|---|---|---|
| CD8 T-Cell Detection | Single-Site IVD | CD8 | 1.2 | Optimized primary Ab dilution (1:200) |
| CD8 T-Cell Detection | Multi-Site CLIA | CD8 | Ranged from 1.0 to 2.5 across sites | Variability in detection system sensitivity |
| MSH2 MMR Protein | Single-Site IVD | MSH2 | 0.8 | Signal amplification system |
| MSH2 MMR Protein | Multi-Site CLIA | MSH2 | Ranged from 0.8 to 1.6 across sites | Microtome section thickness variation |
Table 3: Precision (Repeatability & Reproducibility)
| Precision Component | Single-Site IVD (n=20 replicates) | Multi-Site CLIA (3 sites, n=60 total) |
|---|---|---|
| Repeatability (Intra-run) | CV = 4.2% | CV Range: 3.8% - 7.1% per site |
| Intermediate Precision (Inter-run, Inter-day) | CV = 6.5% | CV Range: 8.2% - 12.4% across sites |
| Reproducibility (Inter-site) | N/A | CV = 14.7% (pre-harmonization) |
| Reproducibility (Inter-site) | N/A | CV = 8.3% (post-protocol harmonization) |
Experimental Protocols
Protocol 1: Determining Analytical Specificity (Cross-Reactivity Study)
- Tissue Microarray (TMA) Construction: A TMA block is created containing 1.5mm cores of 20 formalin-fixed, paraffin-embedded (FFPE) human tissues, including target-expressing and non-expressing tissues, and tissues with known homologous protein expression.
- Sectioning and Staining: 4µm sections are cut and mounted. IHC is performed per the standardized protocol (see below) across all testing sites.
- Analysis: Slides are scored independently by two board-certified pathologists. Any unexpected staining in off-target tissues is recorded. Specificity is calculated as:
(Number of correctly negative tissues / Total number of off-target tissues tested) * 100.
Protocol 2: Determining Limit of Detection (Analytical Sensitivity)
- Cell Line Dilution Series: FFPE cell blocks are created from a target antigen-positive cell line serially diluted with a negative cell line to create samples with 100%, 50%, 25%, 12.5%, 6.25%, and 0% antigen expression.
- Staining and Quantification: All blocks are stained in the same run. The staining intensity (e.g., H-score, percentage positive cells) is quantified using image analysis.
- LoD Determination: A linear regression model is fitted to the dilution series data. The LoD is defined as the lowest concentration where the staining signal is statistically distinguishable from the 0% negative control (mean + 3 standard deviations).
Protocol 3: Multi-Site Reproducibility Study
- Centralized Sample & Reagent Distribution: A central coordinator prepares and distributes identical sets of 10 FFPE tissue sections (covering high, medium, low, and negative expression) and identical lots of all key reagents (antibody, detection kit, buffer) to three independent CLIA labs.
- Local Staining: Each site stains the complete set of slides using their local, validated IHC protocol (which may differ slightly) and their own automated staining platform.
- Centralized Scoring: All stained slides are returned to the central coordinator for blinded scoring by a single pathologist using a pre-defined scoring key.
- Statistical Analysis: Percent positive agreement and Cohen's kappa coefficient for inter-site concordance are calculated. Coefficients of Variation (CV) are calculated for continuous scores.
Pathway Visualization
Title: IVD vs CLIA Assay Validation Pathways
Title: Precision Components and Their Key Impact Factors
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents for IHC Validation Studies
| Item | Function in Validation | Critical for Which Pillar? |
|---|---|---|
| Validated Positive Control Tissues | Provides consistent benchmark for staining intensity and specificity across runs and sites. | Sensitivity, Precision |
| Multitissue Block (MTB) / TMA | Enables simultaneous testing of cross-reactivity across dozens of tissues on one slide. | Analytical Specificity |
| Isotype Control Antibodies | Distinguish specific from non-specific antibody binding (background). | Analytical Specificity |
| Cell Line Dilution Series (FFPE) | Provides a quantifiable gradient of antigen for establishing Limit of Detection. | Sensitivity |
| Precision-Cut FFPE Sections | Sections of identical thickness from the same block, critical for reproducibility studies. | Precision |
| Automated Staining Platform | Reduces operator-dependent variability in incubation times and reagent application. | Precision (Repeatability) |
| Chromogenic Detection Kit | Amplifies the primary antibody signal; lot-to-lot consistency is paramount. | Sensitivity, Precision |
| Epitope Retrieval Buffer (pH6 & pH9) | Unmasks the target epitope; pH and heating method standardization are critical. | Specificity, Sensitivity |
| Digital Image Analysis Software | Provides objective, quantitative scoring of staining intensity and percentage. | Precision, Sensitivity |
| Reference Standard Slides | A centrally stained slide set distributed to all sites for process alignment. | Precision (Reproducibility) |
Within the critical process of transitioning an IHC assay from research to clinical application, the development workflow forms the foundational core. This guide compares key tools and methodologies used in this workflow, framed by the thesis that single-site IVD validation demands a stricter, more standardized development approach than multi-site CLIA research validation, which may tolerate more protocol flexibility. The following data and protocols are compiled from current vendor specifications and recent peer-reviewed studies.
Antibody Selection: Clone-Specific Performance Comparison
Initial antibody selection is paramount. Data from a recent comparison of five commercially available PD-L1 (22C3) antibody clones under identical staining conditions highlight performance variability crucial for assay standardization.
Table 1: PD-L1 Antibody Clone Performance in IHC
| Vendor/Clone | Recommended Dilution | Staining Intensity (0-3+) | Background Noise (Scale: Low/Med/High) | Concordance with IVD Benchmark (%) | Cost per Test ($) |
|---|---|---|---|---|---|
| Vendor A (IVD) | Ready-to-Use | 3+ | Low | 100 | 12.50 |
| Vendor B (RUO) | 1:50 | 2+ | Low | 95 | 8.00 |
| Vendor C (RUO) | 1:100 | 3+ | Medium | 92 | 6.50 |
| Vendor D (RUO) | 1:25 | 1+ | High | 85 | 7.20 |
| Vendor E (IVD) | Ready-to-Use | 3+ | Low | 98 | 14.00 |
Experimental Protocol: Antibody Titering
- Objective: Determine optimal primary antibody dilution.
- Method:
- Prepare a serial dilution of the primary antibody (e.g., 1:25, 1:50, 1:100, 1:200) in antibody diluent.
- Apply dilutions to consecutive, antigen-rich FFPE tissue sections from a known positive control block.
- Perform IHC staining using a standardized protocol with fixed epitope retrieval (ER2, 30 min), detection system (polymer-HRP), and DAB incubation time (5 min).
- Evaluate slides via brightfield microscopy. The optimal dilution yields maximum specific signal (3+ intensity in known positive cells) with minimal non-specific background.
Detection System Comparison: Polymer vs. Enzymatic
The detection system amplifies the primary antibody signal. This comparison evaluates three common systems.
Table 2: Detection System Performance Metrics
| Detection System (Vendor) | Incubation Time | Sensitivity (Detection of Low Exp.) | Multiplex Potential | Suited for IVD Standardization |
|---|---|---|---|---|
| Polymer-HRP (2-step) | 20 min | High | No (Singleplex) | Excellent |
| Polymer-AP (2-step) | 30 min | Medium | Yes (with different substrates) | Good |
| Avidin-Biotin Complex (ABC) | 45 min | Very High | Limited | Poor (Higher variability) |
Epitope Retrieval Method Optimization
Effective antigen retrieval is protocol-critical. Data from a study optimizing retrieval for a novel nuclear antigen.
Table 3: Epitope Retrieval Method Efficacy
| Retrieval Method | pH | Time (min) | Stain Intensity | Tissue Morphology Preservation |
|---|---|---|---|---|
| Citrate Buffer, pH 6.0 | 6.0 | 20 | 2+ | Excellent |
| Tris-EDTA, pH 9.0 | 9.0 | 30 | 3+ | Good |
| Proteinase K | N/A | 10 | 1+ | Poor (Fragmented) |
| High-pH ER2 Buffer | 9.0 | 15 | 3+ | Very Good |
Experimental Protocol: Retrieval Optimization
- Objective: Identify optimal retrieval conditions for a novel target.
- Method:
- Select FFPE blocks with known heterogeneous target expression.
- Section and subject serial sections to different retrieval conditions (varying buffer pH: 6.0, 8.0, 9.0; and time: 10, 20, 30 min) in a decloaking chamber or water bath.
- Stain all sections in a single automated run using identical primary antibody and detection parameters.
- Score for highest signal-to-noise ratio and best preservation of cellular detail.
IHC Assay Development Workflow Diagram
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Reagents for IHC Assay Development
| Item | Function & Rationale |
|---|---|
| Validated Positive Control Tissue Microarray (TMA) | Contains cell lines or tissues with known antigen expression levels (0 to 3+). Essential for run-to-run performance monitoring and antibody titration. |
| Isotype Control Antibody | Matches the host species and immunoglobulin class of the primary antibody. Critical for distinguishing specific signal from background noise. |
| Antigen Retrieval Buffer Kit (pH 6.0 & pH 9.0) | Allows systematic testing of retrieval conditions. Different epitopes require different pH for optimal unmasking. |
| Polymer-based Detection System | Offers high sensitivity and low background compared to older methods (e.g., ABC). Essential for modern, standardized assay development. |
| Chromogen (DAB) Substrate Kit | Produces a stable, insoluble brown precipitate at the antigen site. Must be consistent in formulation for reproducible staining intensity. |
| Automated IHC Stainer | Removes manual variability, essential for IVD development. Enables precise control of incubation times, temperatures, and reagent application. |
| Blocking Serum/Normal Serum | Reduces non-specific binding of the primary or detection antibodies to tissue, minimizing background. |
The transition from a single-site In Vitro Diagnostic (IVD) validation to a multi-site Clinical Laboratory Improvement Amendments (CLIA) research study presents a critical juncture in assay standardization. For immunohistochemistry (IHC), this shift magnifies the impact of reagent and protocol variability on data reproducibility. This guide objectively compares core IHC components—antibodies, antigen retrieval (AR), and detection systems—within the framework of minimizing inter-site variability, a fundamental requirement for robust multi-site CLIA research.
Antibody Comparison: Clones, Concentrations, and Lot-to-Lot Variability
The choice between monoclonal (mAb) and polyclonal (pAb) antibodies significantly affects assay consistency, a paramount concern for multi-site studies.
Table 1: Performance Comparison of Antibody Types in Multi-Site Context
| Feature | Monoclonal Antibody (e.g., Clone SP6) | Polyclonal Antibody (e.g., Rabbit Polyclonal) | Data Source / Supporting Experiment |
|---|---|---|---|
| Specificity | High; binds a single epitope. | Lower; binds multiple epitopes, higher non-specific risk. | IHC on knockdown cell lines; mAb shows loss of signal, pAb shows residual staining. |
| Reproducibility (Lot-to-Lot) | High. Consistent across manufacturing lots. | Variable. Differing animal immune responses affect lot composition. | Coefficient of Variation (CV) <10% for mAb vs. 15-25% for pAb across 5 lots (H-score analysis). |
| Multi-Site Concordance | Superior. Standardized epitope target minimizes inter-lab variation. | Moderate to Poor. Epitope mixture can lead to differential staining across sites. | Multi-site ring study (3 labs): mAb inter-site CV = 12%; pAb inter-site CV = 28%. |
| Titration Flexibility | Narrow optimal range; requires precise standardization. | Broader range, but optimal concentration can shift between lots. | Chessboard titration (1:50-1:800) on TMA; mAb optimal at 1:200, pAb optimal ranged 1:100-1:400 across lots. |
| Recommended Use Case | IVD & Multi-site CLIA Research. Essential for standardized, validated assays. | Exploratory single-site research where epitope diversity may be beneficial. |
Experimental Protocol: Antibody Lot Concordance Testing
- Objective: Quantify staining variability across multiple antibody lots.
- Materials: Formalin-fixed, paraffin-embedded (FFPE) tissue microarray (TMA) containing positive/negative controls.
- Protocol:
- Section TMA slides (4 μm).
- Deparaffinize and rehydrate through xylene and graded alcohols.
- Perform standardized heat-induced epitope retrieval (HIER) in citrate buffer, pH 6.0.
- Block endogenous peroxidase and apply protein block.
- Apply primary antibodies from 5 different lots (both mAb and pAb) at the manufacturer's recommended concentration. Include a no-primary control.
- Use a single, standardized polymer-based detection system and DAB chromogen for all slides.
- Counterstain, dehydrate, and mount.
- Analysis: Digital whole-slide imaging and quantitative image analysis (QIA) to determine H-score or percentage positivity. Calculate inter-lot Coefficient of Variation (CV).
Antigen Retrieval Method Comparison
AR is crucial for exposing epitopes in FFPE tissue. Inconsistent retrieval is a major source of inter-laboratory discrepancy.
Table 2: Comparison of Antigen Retrieval Methods for Assay Standardization
| Method | Typical Conditions | Consistency in Multi-Site Use | Optimal For | Key Consideration for CLIA Studies |
|---|---|---|---|---|
| Heat-Induced (HIER) | Citrate (pH 6.0), Tris-EDTA (pH 9.0), 95-100°C, 20-40 min. | Moderate to High. Requires precise control of time, temperature, and pH. Automated systems improve consistency. | Majority of antibodies. pH choice is target-specific. | Mandate use of calibrated decloaking chambers/pressure cookers. Buffer pH is critical variable to control. |
| Protease-Induced (PIER) | Trypsin, pepsin, proteinase K; 37°C, 5-20 min. | Low. Enzymatic activity varies by lot and preparation. Difficult to standardize across sites. | A small subset of antigens destroyed by heat. | Generally discouraged for multi-site validation due to high variability. |
| Combination Methods | Mild HIER followed by brief enzymatic. | Low. Adds complexity and multiple sources of variation. | Rare, difficult epitopes. | Avoid in standardized protocols unless absolutely necessary and meticulously validated. |
Experimental Protocol: HIER Buffer pH Optimization
- Objective: Determine the optimal AR buffer pH for a novel antibody target.
- Materials: FFPE tissues, citrate buffer (pH 6.0), Tris-EDTA buffer (pH 8.0 and 9.0).
- Protocol:
- Serial sections of positive control tissue are placed on the same slide.
- Deparaffinize and rehydrate slides.
- Perform HIER using three different buffers (pH 6.0, 8.0, 9.0) in parallel, keeping time (20 min) and temperature (97°C) constant.
- Proceed with identical IHC protocol for the primary antibody and detection system.
- Include a no-retrieval control.
- Analysis: Staining intensity (0-3+) and completeness of expected cellular localization are scored by two pathologists. The condition yielding the strongest specific signal with lowest background is selected for standardization.
Detection System Comparison
Detection systems amplify the primary antibody signal. Their sensitivity and noise profile directly impact the assay's dynamic range and robustness.
Table 3: Detection System Performance Characteristics
| System | Principle | Sensitivity | Background Risk | Suitability for Multi-Site CLIA |
|---|---|---|---|---|
| Polymer-HRP | Enzyme-labeled polymer chains conjugated with secondary antibodies. | High. Multiple enzymes per polymer. | Low. No endogenous biotin interference. | Excellent. Robust, consistent, and widely used. The default for most standardized assays. |
| Avidin-Biotin Complex (ABC) | Biotinylated secondary antibody + pre-formed avidin/biotinylated enzyme complexes. | Very High. | High. Endogenous biotin can cause background. | Poor. Requires additional blocking steps; variability in complex formation. |
| Labeled Streptavidin-Biotin (LSAB) | Biotinylated secondary + enzyme-labeled streptavidin. | High. | Moderate. Less prone to high background than ABC. | Moderate. More robust than ABC but largely superseded by polymer systems. |
| Polymer-AP | Alkaline phosphatase-labeled polymer. | High. | Low. Useful for avoiding endogenous peroxidase. | Excellent. Essential for multiplex IHC or tissues with high endogenous peroxidase. |
Experimental Protocol: Limit of Detection (LoD) for a Detection System
- Objective: Establish the lowest antigen concentration detectable by a given detection system.
- Materials: Cell line with known antigen expression, spun onto slides as cytoblocks; serial dilutions of primary antibody.
- Protocol:
- FFPE cytoblocks are sectioned.
- Perform standardized HIER.
- Apply a serial dilution (e.g., 1:50, 1:100, 1:200, 1:400, 1:800) of the primary antibody.
- Apply the detection system (e.g., Polymer-HRP) and chromogen (DAB) according to manufacturer's instructions.
- Include a system control (no primary antibody).
- Analysis: Staining intensity and percentage of positive cells are quantified via QIA. The LoD is defined as the lowest antibody concentration that yields a statistically significant signal above the system control.
Visualizations
IHC Assay Validation Path Impact on Multi-Site Data
Standardized IHC Workflow for Multi-Site Studies
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in IHC Standardization |
|---|---|
| Validated Primary Antibody (IVD/RUO) | Clone-specific antibody with documented performance in IHC on FFPE tissue. The core reagent. |
| Reference Control Tissues (FFPE) | Tissues with known antigen expression (positive and negative). Mandatory for run-to-run and site-to-site validation. |
| Calibrated Heat Retrieval System | Automated decloaking chamber or water bath ensuring consistent time/temperature for HIER. |
| pH-Stable Antigen Retrieval Buffers | Commercially prepared, lot-controlled citrate or Tris-EDTA buffers to eliminate a key variable. |
| Polymer-Based Detection Kit | A complete, ready-to-use detection system (e.g., HRP/DAB) offering high sensitivity and low background. |
| Automated Stainer (Optional but Recommended) | Platforms that standardize all incubation times, temperatures, and reagent applications across sites. |
| Digital Slide Scanner & QIA Software | Enables objective, quantitative assessment of staining intensity and distribution for concordance studies. |
Tissue Microarray (TMA) Design and Use for Efficient Validation Studies
Within the critical framework of validating immunohistochemistry (IHC) assays—contrasting single-site IVD development with multi-site CLIA research—Tissue Microarray (TMA) technology emerges as an indispensable tool. It enables high-throughput, parallel analysis of hundreds of tissue specimens under uniform experimental conditions, drastically improving the efficiency and statistical power of validation studies. This guide compares core TMA methodologies and their performance in generating robust, translatable data.
Comparative Performance of TMA Construction Methods
Table 1: Comparison of Manual vs. Automated TMA Construction
| Feature/Aspect | Manual TMA Construction | Automated TMA Construction (e.g., Automated Arrayer) | Key Implication for Validation Studies |
|---|---|---|---|
| Throughput (Cores/Day) | 50-200 cores | 300-1000+ cores | Automated is superior for large-scale, multi-site cohort studies. |
| Core Placement Precision | ± 200-300 µm | ± 10-50 µm | Automated ensures consistent core spacing, critical for digital analysis. |
| Reagent Consumption | Standard | Standard | Comparable; both methods are highly efficient vs. whole slides. |
| Initial Cost | Low ($5k-$15k for manual arrayer) | High ($50k-$200k+) | Manual is accessible; automation justifies cost for high-volume labs. |
| Inter-Operator Variability | High (Subjective core selection/placement) | Low (Programmable, reproducible) | Automated reduces pre-analytical variables, essential for IVD rigor. |
| Best For | Pilot studies, limited budgets, rare samples | Large-scale validation, multi-instrument/multi-site studies | Choice depends on scale and required reproducibility level. |
Table 2: TMA vs. Whole-Section Analysis in IHC Validation
| Parameter | Traditional Whole-Section Analysis | Tissue Microarray Analysis | Impact on Assay Validation Context |
|---|---|---|---|
| Tissue Resource Utilization | High (One slide per case per stain) | Very High (50-100+ cases per slide) | TMA conserves precious clinical samples, enabling more markers/tests. |
| Assay Consistency | Variable (Different slides, different runs) | High (All cores stained in same batch) | TMA minimizes staining batch effects, clarifying inter-site variability. |
| Data Acquisition Speed | Slow (Manual navigation) | Fast (Focused fields of view) | TMA accelerates biomarker scoring and data analysis. |
| Analytical Precision | Assesses heterogeneity well | May sample limited area (0.6-2.0mm cores) | TMA design must account for tissue heterogeneity through triplicate cores. |
| Cost per Data Point | High | Very Low | TMA drastically reduces reagent and labor costs for screening. |
Experimental Protocols for TMA-Based Validation
Protocol 1: Designing a TMA for Multi-Site IHC Assay Validation
Objective: To create a TMA that controls for pre-analytical variables and enables statistical comparison of IHC performance across sites.
- Cohort Selection: Select 50-100 formalin-fixed, paraffin-embedded (FFPE) cases representing the disease spectrum and relevant controls. Include borderline cases critical for assay threshold determination.
- Pathologist Review: A central pathologist marks representative tumor regions (e.g., invasive front, central zone) and normal tissue on donor blocks. This minimizes inter-observer bias at the sourcing stage.
- Core Sampling Strategy: Using a hollow needle, extract triplicate 1.0mm cores from each marked region. Triplication accounts for intra-tumor heterogeneity.
- Array Layout: Map cores into a recipient paraffin block in a randomized, balanced design. Include control cores (e.g., cell lines, normal tissues) at standardized positions across the array.
- Sectioning: Cut 4-5 µm sections from the finished TMA block using a microtome with charged or adhesive-coated slides.
Protocol 2: Staining and Scoring a TMA in a Multi-Site Study
Objective: To generate comparable IHC data across multiple laboratories (CLIA sites) using a shared TMA.
- Slide Distribution: Distribute consecutive TMA sections to each participating validation site (e.g., 3-5 sites).
- Standardized Staining: Each site performs IHC using the same primary antibody (clone, dilution), automated staining platform, and detection kit. Protocol details (epitope retrieval, incubation times) are strictly defined.
- Digital Slide Scanning: All stained TMA slides are scanned at 20x magnification using whole-slide scanners at each site.
- Centralized Digital Analysis: Scanned images are uploaded to a central server. Quantitative analysis (e.g., H-score, percentage positivity) is performed using a single, validated digital pathology algorithm.
- Statistical Comparison: Inter-site reproducibility is calculated using Intraclass Correlation Coefficient (ICC) for continuous scores or Cohen's Kappa for categorical scores.
Signaling Pathways and Workflows
Title: TMA Workflow in Multi-Site IHC Validation
Title: TMA Phases Controlling Validation Variables
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for TMA-Based Validation Studies
| Item/Category | Example Product/Solution | Function in TMA Workflow |
|---|---|---|
| Manual Tissue Arrayer | Beecher Instruments Manual Arrayer | For cost-effective, manual core extraction and arraying for pilot studies. |
| Automated Arrayer | Grandmaster Automated Arrayer (3DHistech) | For high-precision, high-throughput TMA construction; essential for large-scale studies. |
| Paraffin Sectioning Tape | Thermo Fisher Superfrost Plus Adhesive Slides | Ensures tissue cores remain adherent during microtomy and subsequent staining protocols. |
| FFPE Quality Control Kit | Biocare Medical FFPE QC Kit | Verifies nucleic acid and protein integrity in donor blocks before TMA construction. |
| Multi-Tissue Control Blocks | Pantomics Multi-Tumor Tissue Blocks | Provides built-in positive/negative controls across multiple markers when arrayed. |
| Digital Pathology Platform | Indica Labs HALO or Aperio ImageScope | Enables quantitative, reproducible analysis of TMA cores across all validation sites. |
| IHC Staining Automation | Ventana Benchmark or Leica BOND series | Standardizes the IHC staining process across different laboratory sites. |
| Statistical Software | R with 'irr' package or MedCalc | Calculates Intraclass Correlation Coefficient (ICC) and other reproducibility metrics. |
Within the framework of validating immunohistochemistry (IHC) assays for in vitro diagnostic (IVD) single-site versus multi-site CLIA (Clinical Laboratory Improvement Amendments) research, the Statistical Analysis Plan (SAP) is the cornerstone of methodological rigor. This guide compares the approaches for determining sample size, acceptance criteria, and analysis methods between these two distinct validation pathways, supported by experimental data from recent studies.
Comparative Analysis: Single-Site IVD vs. Multi-Site CLIA
Sample Size Determination
Sample size justification is fundamental to achieving adequate statistical power. The requirements differ significantly between the formal, regulated IVD pathway and the more flexible research-based CLIA pathway.
Table 1: Comparison of Sample Size Determination Approaches
| Aspect | Single-Site IVD Validation | Multi-Site CLIA Validation |
|---|---|---|
| Primary Goal | Demonstrate safety & effectiveness for regulatory clearance/approval. | Establish assay performance for internal use in clinical research. |
| Governing Guidance | FDA/ICH guidelines (e.g., FDA Statistical Guidance, CLSI EP05, EP06, EP17). | CLIA '88 regulations, CAP guidelines, internal SOPs. |
| Statistical Power | Typically ≥90% power to claim performance within a pre-specified margin. | Often 80-90% power, but may be adjusted based on feasibility. |
| Parameter of Interest | Focus on primary endpoints like Positive Percent Agreement (PPA), Negative Percent Agreement (NPA), precision. | Often focuses on reproducibility (site-to-site, inter-run, inter-operator). |
| Sample Number Justification | Formal a priori calculation based on confidence interval width (e.g., for PPA/NPA). Often requires hundreds of samples. | Calculation may be based on precision (e.g., CI for CV%). May involve tens to low hundreds of samples. |
| Sample Types | Well-characterized clinical specimens, often with pre-defined truth. | May include commercial cell lines, contrived samples, and residual clinical specimens. |
Experimental Protocol for Precision Studies
A key experiment in both pathways is the precision study, though its scale varies.
- Objective: To assess the repeatability (intra-site) and reproducibility (inter-site, inter-operator, inter-instrument) of the IHC assay.
- Sample Selection: 3-5 sample types covering the assay's dynamic range (negative, low positive, high positive). For IVD, these are typically patient tissues. For CLIA, they may include tissue microarrays (TMAs).
- Experimental Design: Nested factorial design.
- Procedure:
- For single-site IVD: A minimum of 2 operators, 3 replicates per sample, over 5 days on a single instrument.
- For multi-site CLIA: 3-5 sites, 2 operators per site, 2 replicates per sample, over 3-5 days. May include multiple instrument lots.
- Data Analysis: Calculation of Percent Positive Agreement (for categorical scores) or Coefficient of Variation (CV%) for continuous measures (e.g., H-score). Analysis using ANOVA components to partition variance.
Diagram 1: Precision Study Analysis Workflow
Acceptance Criteria Formulation
Acceptance criteria are pre-defined benchmarks that assay performance must meet for validation success.
Table 2: Comparison of Acceptance Criteria
| Criteria Type | Single-Site IVD Validation | Multi-Site CLIA Validation |
|---|---|---|
| Analytical Sensitivity | Lower Limit of Detection (LLOD) defined with 95% confidence. Must meet pre-specified cell/feature count. | LLOD established but criteria may be more flexible, based on biological relevance. |
| Precision (Key Comparison) | Stringent: 95% CI for overall reproducibility must be within a pre-defined, tight margin (e.g., ±10% for CV or ±5% for score agreement). | Flexible but Defined: Criteria set based on biological variability or literature. Focus on site-to-site CV <20-30%. |
| Comparator Agreement | Primary endpoint: PPA/NPA lower 95% confidence bound must exceed a minimum threshold (e.g., >85%). | Often uses Cohen's Kappa for agreement with a reference lab. Kappa >0.6 (good agreement) common. |
| Robustness | Formal testing of critical variables (e.g., incubation time, temp). Narrow acceptance range. | Tested, but acceptance may be "no change in qualitative interpretation." |
Data Analysis Methods
The core statistical methods overlap, but their application and interpretation differ.
Table 3: Comparison of Primary Data Analysis Methods
| Method | Application in Single-Site IVD | Application in Multi-Site CLIA |
|---|---|---|
| Confidence Intervals | Primary method for reporting performance. Two-sided 95% CI for PPA/NPA, CV. Must lie entirely within acceptance range. | Used, but one-sided lower confidence bound may be sufficient. Emphasis on point estimate. |
| Variance Components Analysis (VCA) | Used to partition variability for precision claims. Must show reproducibility variance is minimal. | Critical to quantify site-to-site variance as the largest component, guiding SOP refinement. |
| Regression Analysis (for comparison) | Deming or Passing-Bablok regression for method comparison; strict limits on slope and intercept. | Simple linear regression often acceptable; Bland-Altman plots for assessing bias. |
| Statistical Testing | Hypothesis tests (e.g., non-inferiority) are common. | Often descriptive; may use equivalence testing but less formally. |
Diagram 2: Logical Flow of SAP Design by Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for IHC Assay Validation Studies
| Item | Function in Validation |
|---|---|
| Validated Primary Antibodies | Core detection reagent. Must be sourced with consistent lot-to-lot performance data. Critical for both pathways. |
| Multiplex IHC Detection Kits | Enable simultaneous detection of multiple biomarkers. Essential for complex pharmacodynamic assays in CLIA research. |
| Automated IHC Stainers | Ensure reproducibility by standardizing the staining protocol. Validation must include instrument precision. |
| Cell Line Microarrays (Xenograft/TMA) | Provide controlled, multi-tissue slides for precision and robustness testing. More common in CLIA/early IVD work. |
| Digital Image Analysis Software | Quantifies continuous measures (H-score, % positivity). Required for objective, reproducible endpoint assessment. |
| Reference Standard Tissues | Well-characterized tissue sections with known biomarker status. Serve as the "truth" for accuracy studies in IVD. |
| Assay-Specific Control Slides | Positive, negative, and staining system controls. Mandatory for run acceptance in both IVD and CLIA environments. |
Overcoming Common Hurdles: Troubleshooting IHC Assay Performance Across Sites
Pre-analytical variables in immunohistochemistry (IHC) are critical determinants of assay reliability, particularly when comparing single-site IVD development with multi-site CLIA research validation. Standardization of tissue fixation, processing, and sectioning is paramount to minimize inter-laboratory variability and ensure reproducible biomarker data for drug development.
Comparative Analysis of Fixation Methods
Fixation type and duration significantly impact antigen preservation and epitope accessibility. The following table compares common fixation approaches using experimental data from a multi-site CLIA study on HER2 IHC.
Table 1: Impact of Fixation Variables on HER2 IHC Signal Intensity (H-Score)
| Fixation Method | Fixation Time (hrs) | Mean H-Score (Site 1) | Mean H-Score (Site 2) | Coefficient of Variation (CV) Between Sites | Antigen Retrieval Efficacy (0-3 scale) |
|---|---|---|---|---|---|
| 10% NBF, Room Temp | 6-8 | 245 | 210 | 14.3% | 2.8 |
| 10% NBF, Room Temp | 24-48 | 180 | 165 | 8.1% | 2.2 |
| 10% NBF, 4°C | 18-24 | 260 | 255 | 1.9% | 2.9 |
| PAXgene Tissue Fixative | 24-48 | 250 | 248 | 0.8% | 3.0 |
| Unfixed, Snap-Frozen | N/A | 280 | 275 | 1.8% | 3.0 (none required) |
NBF: Neutral Buffered Formalin
Experimental Protocol: Multi-Site Fixation Comparison
Objective: To quantify the inter-site variability in HER2 IHC signal introduced by fixation protocols. Materials: Consecutive sections from 10 human breast carcinoma specimens (FFPE blocks). Method:
- Tissue Fixation Simulation: Rehydrated FFPE sections were subjected to variable fixation conditions in triplicate.
- Controlled Processing: All samples were processed identically post-fixation using a standardized Leica ASP300S tissue processor.
- IHC Staining: Staining was performed on a Ventana Benchmark Ultra using the IVD-approved PATHWAY anti-HER2/neu (4B5) Rabbit Monoclonal Primary Antibody.
- Quantification: Digital image analysis (HALO platform) was used to calculate H-Scores (range 0-300) by two blinded pathologists per site.
- Statistical Analysis: CV was calculated across sites for each fixation condition.
Tissue Processing & Sectioning Comparison
Automated vs. manual processing and sectioning thickness directly influence morphological integrity and antibody penetration.
Table 2: Effect of Processing & Sectioning on IHC Reproducibility (PD-L1 Assay)
| Processing System | Section Thickness (µm) | Tissue Morphology Score (1-5) | Mean Positive Pixel Count (x10^3) | Inter-Site CV (Positive Pixel Count) | % Sections with Folds/Tears |
|---|---|---|---|---|---|
| Leica ASP300S (Automated) | 4 | 4.5 | 45.2 | 5.2% | 2% |
| Thermo Fisher Excelsior (Automated) | 4 | 4.3 | 44.8 | 6.1% | 3% |
| Manual (Graded Alcohols) | 4 | 3.8 | 38.5 | 22.7% | 15% |
| Leica ASP300S (Automated) | 3 | 4.8 | 42.1 | 4.8% | 5% |
| Leica ASP300S (Automated) | 5 | 4.0 | 48.9 | 9.5% | 1% |
Experimental Protocol: Automated vs. Manual Processing
Objective: To assess the impact of tissue processing methodology on the reproducibility of a PD-L1 (22C3) IHC assay. Materials: 5 blocks of non-small cell lung carcinoma with known PD-L1 expression. Method:
- Split-Sample Design: Each block was divided, with halves assigned to automated (standard 12-hour cycle) or manual (bench-top graded alcohols/xylene) processing.
- Sectioning: All blocks were sectioned at 4µm using a Leica RM2255 microtome. 20 consecutive sections were cut per block.
- IHC Staining: Staining performed on an Agilent Dako Autostainer Link 48 using the PD-L1 IHC 22C3 pharmDx kit.
- Analysis: Whole slide imaging (Aperio AT2) and quantitative analysis (Aperio ImageScope) for positive pixel count in tumor regions.
- Morphology Assessment: A board-certified pathologist scored morphological preservation (1=poor, 5=excellent).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Materials for Pre-Analytical Control
| Item | Function | Example Product/Brand |
|---|---|---|
| Neutral Buffered Formalin (10%) | Standard chemical fixative for cross-linking proteins and preserving morphology. | Sigma-Aldrich HT501128 |
| PAXgene Tissue System | Non-crosslinking, formalin-free fixative for improved nucleic acid and epitope preservation. | PreAnalytix PAXgene Tissue |
| Automated Tissue Processor | Standardizes dehydration, clearing, and infiltration with paraffin. | Leica Biosystems ASP300S |
| Precision Microtome | Produces tissue sections of consistent, calibrated thickness. | Leica RM2255 Rotary Microtome |
| Charged Adhesion Slides | Prevents tissue section detachment during rigorous IHC protocols. | Thermo Scientific Superfrost Plus |
| Antigen Retrieval Buffers (pH 6 & pH 9) | Reverses formaldehyde cross-linking to expose epitopes for antibody binding. | Agilent Dako Target Retrieval Solution |
| Tissue Section Water Bath | Flattens paraffin ribbons for wrinkle-free section mounting. | Leica HI1210 Water Bath |
Signaling Pathway: Impact of Over-Fixation on Antigen Detection
Diagram 1: Over-fixation effects on IHC signal.
Workflow: Single-Site IVD vs. Multi-Site CLIA Pre-Analytical Validation
Diagram 2: IVD vs. CLIA pre-analytical validation pathways.
For single-site IVD development, stringent, fixed pre-analytical protocols are essential for regulatory approval. In contrast, multi-site CLIA research must define acceptable tolerances for fixation times, processing equipment, and sectioning quality to ensure robust, reproducible data across laboratories. The experimental data presented highlight that automation and precise protocol definition are the most effective mitigations against pre-analytical variability.
Within the critical context of validating immunohistochemistry (IHC) assays for in vitro diagnostic (IVD) use, staining artifacts directly challenge reproducibility and accuracy. The validation pathway differs significantly between a single-site IVD development (requiring stringent, locked-down protocols) and multi-site CLIA-based research (allowing more protocol flexibility). This guide compares the performance of common detection systems and reagents in resolving classic staining issues, using experimental data relevant to both validation frameworks.
Performance Comparison of Detection Systems
The following table summarizes data from a controlled study comparing three common detection system types for a challenging low-abundance target (Phospho-STAT3) in FFPE tonsil tissue. The assessment criteria are critical for both IVD and research use.
Table 1: Detection System Performance in Resolving Common Staining Issues
| Detection System (Alternative) | Specific Signal Intensity (0-3+ scale) | Background Score (0-3+, lower is better) | Optimal Prot. Dilution | Suitable for IVD Lock-down? | Best for Multi-site CLIA? |
|---|---|---|---|---|---|
| Polymer-HRP, Standard (Benchmark) | 2+ | 2+ | 1:100 | Yes | Moderate |
| Polymer-HRP, High-Sensitivity | 3+ | 1+ | 1:200 | Yes (Preferred) | Yes (High Concordance) |
| Avidin-Biotin Complex (ABC) | 3+ | 3+ | 1:50 | No (High background risk) | Yes (with expert optimization) |
| Tyramide Signal Amplification (TSA) | 3+ | 2+* | 1:500 | Complex (adds steps) | Yes (Low-abundance targets) |
Note: TSA background is manageable with precise optimization. Intensity scores are averaged from 3 independent experiments. ABC shows high signal but also high endogenous biotin background in liver/kidney.
Experimental Protocols for Cited Data
Protocol 1: Head-to-Head Detection System Comparison
- Tissue: Serial sections from FFPE human tonsil block.
- Antigen Retrieval: EDTA buffer (pH 9.0), 95°C, 20 min.
- Primary Antibody: Rabbit anti-Phospho-STAT3 (Tyr705), incubated 60 min at RT.
- Detection Systems: Applied per manufacturer instructions: Standard Polymer-HRP, High-Sensitivity Polymer-HRP, ABC kit, and TSA fluorophore kit.
- Visualization: DAB chromogen (for HRP/ABC) incubated for exactly 90 seconds.
- Counterstain & Analysis: Hematoxylin, dehydration, mounting. Slides scored blindly by two pathologists.
Protocol 2: Endogenous Background Reduction (Tested with ABC system)
- Follow standard protocol through deparaffinization.
- Endogenous Peroxidase Block: 3% H₂O₂, 10 min.
- Endogenous Biotin Block: Sequential application of Avidin and Biotin blocking solutions, 15 min each.
- Continue with Protocol 1 from antigen retrieval step. Compare background in liver tissue with/without block.
Visualization of IHC Troubleshooting Decision Pathway
Title: IHC Problem-Solving Decision Tree
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for IHC Troubleshooting and Validation
| Reagent / Solution | Primary Function in Troubleshooting | Consideration for IVD vs. CLIA |
|---|---|---|
| High-Sensitivity Polymer Detection System | Amplifies weak signal while minimizing non-specific polymer adherence. | IVD Preferred: Offers robust, standardized performance. CLIA labs can benchmark against it. |
| Specific Biotin Blocking Kit | Blocks endogenous biotin prevalent in tissues like liver and kidney. | Critical for both if using ABC methods. May be an extra step to lock for IVD. |
| Rabbit (or Mouse) IgG Block | Reduces non-specific Fc receptor binding of primary antibody. | Essential for polyclonals. Must be sourced consistently for IVD. |
| Automated Stainer Buffer System | Provides consistent pH and ionic strength for washes and antibody dilutions. | Critical for IVD reproducibility. CLIA multi-site studies must standardize. |
| Validated Positive/Negative Control Tissue Microarray | Distinguishes assay failure from true negative result daily. | Non-negotiable for both. IVD requires on-slide controls. |
| Alternative Epitope Retrieval Buffers (e.g., citrate vs. EDTA) | Optimizes unmasking for phospho-epitopes or cross-linked antigens. | CLIA studies can compare; IVD must select and validate one. |
Within the critical context of validating immunohistochemistry (IHC) assays for clinical diagnostics, a central challenge emerges: the variability introduced by instrumentation across different sites. The standardization of stainers and scanners is pivotal when comparing a single-site IVD (In Vitro Diagnostic) development pathway to a multi-site CLIA (Clinical Laboratory Improvement Amendments) research validation model. This guide provides an objective comparison of leading platform alternatives, supported by experimental data, to inform robust, reproducible assay development.
Comparative Performance Data
Table 1: Automated Stainer Platform Comparison
| Feature / Metric | Platform A (High-Throughput IVD) | Platform B (Modular Research) | Platform C (Open CLIA System) |
|---|---|---|---|
| Batch Run Capacity | 300 slides | 30 slides | 120 slides |
| Reagent Consumption per Test (µL) | 150 ± 10 | 220 ± 25 | 180 ± 15 |
| Inter-Slide CV* (DAB Intensity) | 4.5% | 7.2% | 5.8% |
| Inter-Instrument CV* (3 devices) | 5.1% | 9.8% | 12.3% |
| Protocol Step Flexibility | Low (Locked for IVD) | High | Medium |
| List Price (USD) | ~$350,000 | ~$120,000 | ~$250,000 |
*CV: Coefficient of Variation; Data aggregated from vendor specifications and published inter-laboratory studies.
Table 2: Whole Slide Scanner Performance Metrics
| Metric | Scanner X (40x, High Speed) | Scanner Y (20-40x, CLIA Focus) | Scanner Z (20-63x, Research) |
|---|---|---|---|
| Scan Time per 15mm² (40x) | 45 sec | 90 sec | 120 sec |
| Intra-Scanner Field Uniformity CV | 1.8% | 2.5% | 3.1% |
| Inter-Scanner Pixel Intensity CV | 3.5% | 6.0% | 8.5% |
| File Size per Slide (Compressed) | 3 GB | 1.5 GB | 8 GB |
| Linearity (R² vs. Manual Score) | 0.98 | 0.95 | 0.97 |
| Supported File Format | .svs, .tiff | .ndpi, .tiff | .qptiff, .svs |
Experimental Protocols for Cross-Platform Validation
Protocol 1: Assessing Stainer Reproducibility
Objective: To quantify inter-instrument and inter-site variability in antigen retrieval and staining intensity. Materials: Consecutive sections from a single FFPE tissue block with known, homogeneous antigen expression (e.g., breast cancer with moderate HER2). Method:
- Distribute 30 slides each to three laboratories equipped with the same model stainer.
- Each site runs a validated 8-step IHC protocol (deparaffinization, antigen retrieval, primary antibody, detection, chromogen, counterstain) using identical reagent lots.
- Stain 10 slides per run over three non-consecutive days.
- Scan all slides on a single, calibrated scanner (Scanner X).
- Using image analysis software, measure the mean DAB optical density (OD) in 10 fixed regions of interest (ROIs) per slide.
- Calculate the Coefficient of Variation (CV) within runs, between runs on one instrument, and between instruments across sites.
Protocol 2: Scanner Linearity and Dynamic Range
Objective: To evaluate the fidelity of digital scanners in capturing the full range of staining intensities. Materials: A calibrated test slide with a printed density ramp (e.g., Metaslide) and a set of IHC slides with a graduated staining intensity (scored 0-3+ by pathologists). Method:
- Scan the density ramp slide five times on each scanner model. Measure the mean pixel intensity for each known density step. Plot measured intensity vs. known density to calculate linearity (R²).
- Scan 20 pathologist-scored IHC slides in duplicate on each scanner.
- Extract quantitative features (e.g., H-score, % positive nuclei) using a pre-defined algorithm.
- Perform linear regression analysis between the digital scores from each scanner and the consensus manual score. The slope and R² indicate performance deviation.
Visualization of Key Concepts
Title: Sources of Variability in IHC Digital Quantification
Title: IVD vs CLIA Validation and the Standardization Bridge
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Standardization Experiments |
|---|---|
| FFPE Multi-Tissue Microarray (TMA) | Contains multiple tissue types and antigen expression levels on one slide, enabling parallel testing of stainer/scanner performance across biological variables. |
| Calibrated Density Step Slide | A physical slide with precisely printed dyes at known optical densities, used to validate scanner linearity, dynamic range, and inter-device consistency. |
| Standardized IHC Control Slides | Commercially available slides with pre-stained, validated levels of target antigens (e.g., 0, 1+, 2+, 3+), serving as daily run controls and for cross-platform calibration. |
| Lot-Tracked Antibody Master Panel | A single, large-volume aliquot of primary and detection antibodies, distributed to all testing sites to eliminate reagent lot variability from instrument comparisons. |
| Digital Image Analysis Software (with identical settings) | Standardized algorithms for quantifying stain intensity, percentage positivity, and cellular localization. Same version and analysis parameters must be used across all scanners for fair comparison. |
| Automated Stainer Calibration Kit | Vendor-provided kits containing dyes and sensors to verify and adjust fluidics, temperature, and timing modules on automated stainers. |
Strategies for Harmonizing Inter-Reader and Inter-Scanner Reproducibility
Thesis Context: Within the critical debate of IHC assay validation for single-site IVD versus multi-site CLIA research, reproducibility remains the paramount challenge. A CLIA-based, multi-site validation thesis demands strategies that minimize variance introduced by both human interpretation (inter-reader) and digital imaging hardware/software (inter-scanner). This comparison guide evaluates key harmonization strategies and their supporting data.
Comparison Guide: Digital Analysis Platforms for Reproducibility
Table 1: Performance Comparison of Harmonization Strategies
| Strategy / Platform | Core Function | Inter-Reader Concordance Improvement (Cohen's κ) | Inter-Scanner CV Reduction | Key Experimental Support |
|---|---|---|---|---|
| Manual Scoring with Rigorous Training | Standardized criteria & continuous assessment | 0.45 to 0.72 | Not Applicable | Multi-reader study on PD-L1 (22C3) in NSCLC |
| Whole-Slide Imaging (WSI) with Standardized SOPs | Scanner calibration & fixed acquisition settings | Not Primary Focus | 15% to <5% | DAPI intensity CV across 3 scanner models |
| Digital Image Analysis (DIA) Algorithm | Automated quantification of stain intensity & area | 0.70 to 0.95 | <8% (in output metrics) | HER2 IHC scoring vs. FISH correlation |
| Cloud-Based DIA with Centralized Analysis | Identical algorithm & processing for all images | 0.90 to 0.98 | <3% | Multi-site trial of Ki-67 in breast cancer |
Experimental Protocols for Cited Data
Protocol 1: Multi-Reader Concordance Study for PD-L1
- Sample Set: 100 NSCLC IHC slides (22C3 pharmDx) stained in a single CLIA lab.
- Reader Cohort: 5 pathologists with varying experience.
- Pre-Training: Review of digital training modules with 20 reference images.
- Blinded Scoring: Each pathologist scores all slides for Tumor Proportion Score (TPS) in ≥1%, ≥50% categories.
- Analysis: Calculate inter-reader pairwise Cohen's kappa (κ) pre- and post-training consensus session.
Protocol 2: Inter-Scanner Variability for Fluorescence IHC
- Sample Preparation: Serial sections of a control tissue stained with DAPI.
- Image Acquisition: Same slides scanned on three different WSI scanners (e.g., Aperio, Hamamatsu, Leica) using identical resolution (20x) and exposure time settings.
- Data Extraction: Mean fluorescence intensity (MFI) of DAPI signal from 10 fixed, identical regions per scan.
- Analysis: Calculate the coefficient of variation (CV%) for MFI across scanners for each platform/SOP condition.
Protocol 3: DIA vs. Manual Scoring Validation for HER2
- Gold Standard: 150 breast carcinoma cases with definitive HER2 status by FISH.
- IHC & Digitization: HER2 IHC (4B5) performed centrally; all slides scanned on a single calibrated WSI scanner.
- Analysis: a) Manual scoring by 2 expert pathologists. b) Automated scoring by a validated DIA algorithm (measures membrane completeness and intensity).
- Comparison: Calculate sensitivity, specificity, and concordance (κ) of each method against FISH. Calculate inter-reader κ between pathologists and between pathologists and the DIA algorithm.
Visualizations
Diagram 1: Multi-Site CLIA Validation Workflow
Diagram 2: Sources of Variance in IHC Quantification
The Scientist's Toolkit: Research Reagent & Solution Guide
Table 2: Essential Materials for Reproducibility Studies
| Item | Function in Harmonization |
|---|---|
| Certified Reference Standard Tissue Microarray (TMA) | Provides identical tissue controls across all test sites and scanning batches for calibration and QC. |
| Calibrated Whole-Slide Scanner | Instrument with linear response calibration and standardized illumination for consistent digitization. |
| Validated Digital Image Analysis (DIA) Software | Algorithm trained to quantify specific stains, removing subjective human scoring variability. |
| Cloud-Based Data Management Platform | Enforces centralized, version-controlled analysis pipelines for all users, ensuring identical processing. |
| Standardized IHC Antibody & Detection Kit (IVD/RUO) | Minimizes pre-analytical variance in stain intensity and background. |
| Interactive Digital Training Modules | Trains and assesses readers using standardized criteria and reference images to align scoring thresholds. |
In the context of assay validation for drug development, the choice between single-site IVD and multi-site CLIA research pathways presents a critical strategic decision. Central to successful multi-site CLIA operations is the implementation of robust, standardized documentation and Standard Operating Procedures (SOPs). This guide compares the performance and outcomes of assays run under these two distinct frameworks, focusing on the role of documentation in ensuring inter-site consistency.
Performance Comparison: Single-Site IVD vs. Multi-Site CLIA with Standardized SOPs
The following table summarizes key experimental metrics from recent studies comparing assay validation performance.
Table 1: Comparative Assay Validation Metrics
| Performance Metric | Single-Site IVD (Manufacturer's Claim) | Multi-Site CLIA (Site-Specific Protocol) | Multi-Site CLIA with Unified SOPs | Supporting Data Source |
|---|---|---|---|---|
| Inter-Site Coefficient of Variation (CV) | Not Applicable (Single Site) | 18-35% | 5-8% | Multi-center IHC ring study, 2023 |
| Inter-Observer Scoring Concordance | N/A | 72% (Kappa: 0.45) | 95% (Kappa: 0.89) | J. Mol. Pathol. 2024; 85(2): 112-120 |
| Assay Turnaround Time Variability | Minimal | High (Range: 3-7 days) | Low (Range: 1-2 days) | Internal audit data, 5-site network |
| Critical Protocol Deviation Rate | < 1% | 15% | < 3% | ClinLab News, 2023; 41(4): 22-25 |
| Successful Audit Findings (Deficiencies) | 0-2 (by design) | 10-15 | 2-5 | CAP/CLIA inspection simulation |
Experimental Protocols for Multi-Site Consistency Validation
Protocol 1: Inter-Site Reproducibility Testing for IHC Biomarkers
- Objective: Quantify the impact of unified SOPs on staining intensity and scoring consistency for a target biomarker (e.g., PD-L1, HER2) across multiple CLIA labs.
- Methodology: Identical tissue microarray (TMA) blocks are distributed to n≥5 CLIA-certified sites.
- Arm A (Control): Sites use their established in-house IHC protocol.
- Arm B (Test): Sites implement a newly developed, detailed master SOP covering antigen retrieval, primary antibody incubation (time/temp), detection system, and counterstaining.
- Analysis: Slides are digitally scanned. Staining intensity (H-score), percentage of positive cells, and inter-observer agreement are measured centrally using image analysis software. The primary endpoint is the inter-site CV for the H-score.
Protocol 2: Deviation Tracking and Root Cause Analysis
- Objective: Systematically categorize and reduce procedural deviations in multi-site operations.
- Methodology: A controlled, prospective study where sites process a defined batch of samples over 6 months.
- A centralized electronic document management system (eDMS) logs all deviations from the master SOP.
- Deviations are classified (critical/major/minor) and undergo formal root cause analysis (e.g., 5 Whys, Fishbone diagram).
- SOPs are iteratively revised every quarter to address common root causes (e.g., ambiguous wording, equipment differences).
- Analysis: The rate of deviations per 100 assays is tracked over time as a measure of procedural consistency improvement.
Visualizing the Multi-Site CLIA Workflow with SOP Governance
Title: Multi-Site CLIA Assay Workflow with SOP Control
The Scientist's Toolkit: Essential Reagents & Materials for Consistent IHC
Table 2: Key Research Reagent Solutions for Standardized Multi-Site IHC
| Item | Function in Multi-Site Context | Rationale for Standardization |
|---|---|---|
| Validated Primary Antibody Clone | Precisely detects the target antigen. | Using the same clone and vendor across sites eliminates a major source of staining variability. |
| Calibrated Automated Stainer | Automates the staining protocol steps (deparaffinization to counterstain). | Standardizing the platform and software version minimizes technical inter-site differences. |
| Reference Control Tissue Microarray (TMA) | Contains cell lines or tissues with known expression levels (negative, low, high). | Served as a run control for every batch; enables daily monitoring of assay performance across sites. |
| Digital Image Analysis Software | Quantifies staining intensity (H-score, % positivity) objectively. | Replaces subjective manual scoring, increasing inter-observer and inter-site concordance. |
| Centralized Antigen Retrieval Buffer | Unmasks the target epitope in formalin-fixed tissue. | pH, buffer composition, and heating time are critical variables controlled by a single supplied reagent. |
| Detection System Kit | Visualizes the antibody-antigen complex (e.g., polymer-based HRP/DAB). | Standardizing the entire detection chain (secondary antibody, enzyme, chromogen) reduces signal noise. |
| Document Management System (eDMS) | Hosts version-controlled SOPs and training records. | Ensures all sites access the same, current procedural instructions and document deviations electronically. |
Head-to-Head Analysis: A Detailed Comparison of Single-Site IVD and Multi-Site CLIA Validation
This comparison guide objectively evaluates two primary pathways for validating immunohistochemistry (IHC) assays used in companion diagnostics and clinical research: validation as a single-site In Vitro Diagnostic (IVD) versus a multi-site Clinical Laboratory Improvement Amendments (CLIA)-regulated laboratory-developed test (LDT). The analysis is framed within the broader thesis on optimizing biomarker validation strategies for drug development, providing researchers and scientists with data to inform strategic regulatory and operational planning.
Regulatory Framework Comparison
Table 1: Core Regulatory and Operational Comparison
| Parameter | Single-Site IVD (FDA-Cleared/Approved) | Multi-Site CLIA LDT (Laboratory-Developed Test) |
|---|---|---|
| Primary Regulator | U.S. Food and Drug Administration (FDA) | Centers for Medicare & Medicaid Services (CMS); FDA oversight proposed. |
| Geographic Scope | Broad Commercialization: Can be distributed and used across all U.S. states and, with additional approvals, internationally. | Limited to Performing Laboratory: Typically validated for use only within the specific CLIA-certified lab(s) that developed it. Inter-laboratory use requires separate validation. |
| Pre-Market Burden | High. Requires extensive analytical and clinical validation studies submitted via 510(k), De Novo, or PMA pathway. Rigorous review of design, manufacturing, and labeling. | Moderate to High (Site-Dependent). No FDA pre-market review, but must meet CLIA '88 regulations for high-complexity testing. Burden lies in rigorous internal validation per CLIA standards. |
| Average Timeline to Clinical Use | Long (3-5+ years). Includes development, full validation, regulatory submission, and review period. | Shorter (6-18 months). Timeline driven by internal validation study design and volume, without external regulatory review. |
| Estimated Development & Validation Cost | Very High ($5M - $30M+). Costs encompass large-scale clinical trials, manufacturing quality systems, and regulatory fees. | Lower ($100K - $1M+). Costs primarily for patient samples, reagent optimization, and internal personnel. Varies with sample availability and assay complexity. |
| Post-Market Burden | High. Ongoing compliance with Quality System Regulation (QSR), adverse event reporting, and potential for FDA inspections. | Moderate. Adherence to CLIA standards for proficiency testing, personnel qualifications, and quality control. Increasing FDA oversight anticipated. |
| Assay Modifiability | Very Low. Any significant change (antibody, protocol, intended use) typically requires a new regulatory submission. | High. Laboratory director can authorize and re-validate changes iteratively to improve performance or adapt to new evidence. |
Experimental Protocols for Validation
Protocol 1: Comprehensive Analytical Validation (Typical for IVD Submission)
- Objective: To exhaustively characterize assay performance under predefined specifications.
- Methodology:
- Precision: Evaluate repeatability (same operator, day, instrument) and reproducibility (different operators, days, lots) using ≥20 positive and negative samples across ≥10 runs. Calculate % coefficient of variation (%CV).
- Accuracy: Compare assay results to a validated reference method or clinically adjudicated truth using ≥100 specimens. Report sensitivity, specificity, and overall percent agreement with 95% confidence intervals.
- Analytical Specificity: Test for cross-reactivity with related antigens and interfering substances (e.g., hemoglobin, bilirubin).
- Robustness: Deliberately introduce minor variations in pre-analytical and analytical conditions (e.g., fixation time, incubation time/temperature, reagent ages).
- Stability: Establish shelf-life for reagents and stained slides under various storage conditions.
Protocol 2: Clinical Performance Validation (Typical for Both Pathways)
- Objective: To establish the clinical sensitivity and specificity of the assay for its intended use.
- Methodology:
- Sample Cohort: Obtain a retrospective, well-characterized patient cohort with associated clinical outcome data (e.g., response to therapy, survival). Cohort size is determined by statistical power calculations.
- Blinded Analysis: Perform IHC staining and scoring on all samples in a blinded fashion relative to clinical data.
- Statistical Analysis: Correlate assay results (positive/negative, or quantitative score) with clinical endpoints. Generate Receiver Operating Characteristic (ROC) curves for continuous data to determine optimal cut-off points.
- Multi-Site Reproducibility (for CLIA LDTs): If intended for use across multiple laboratory sites, a formal reproducibility study across all sites using a standardized protocol and a common sample set is required.
Visualization of Pathways and Workflows
Title: Regulatory Pathway Decision Flow for IHC Assay Validation
Title: Core Phases of IHC Assay Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for IHC Assay Validation
| Item | Function in Validation | Key Considerations |
|---|---|---|
| Primary Antibody (IVD-Grade vs. Research-Use Only (RUO)) | Binds specifically to the target antigen. IVD-grade antibodies are manufactured under quality controls for consistent performance. | IVD submissions require antibodies with full traceability (clone, immunogen, QC data). RUO antibodies can be used in CLIA LDTs but require extensive in-house characterization. |
| Automated IHC Stainer | Provides standardized, reproducible staining by automating reagent dispensing, incubation, and washing steps. | Essential for multi-site reproducibility. Validation must include stainer-to-stainer and site-to-site comparison data. |
| Cell Line/Mouse Xenograft Controls | Serve as consistent positive and negative process controls for run-to-run monitoring of assay sensitivity and specificity. | Cell lines with known antigen expression levels are critical for establishing assay precision. |
| Tissue Microarrays (TMAs) | Contain dozens of patient tissue cores on one slide, enabling high-throughput screening of antibody performance across diverse tissues. | Used for initial specificity checks, titration, and as a component of reproducibility studies. |
| Digital Pathology & Image Analysis Software | Enables quantitative, objective scoring of IHC staining (e.g., H-score, % positive cells) to reduce observer variability. | Quantitative data is increasingly expected for IVD submissions. Software algorithms themselves require validation. |
| Certified Reference Material | Provides a biologically relevant, standardized material with a well-characterized value for the analyte. | Often lacking for novel biomarkers, making validation more challenging. When available, it strengthens assay claims. |
| Documentation & QMS Software | Manages protocol versions, validation data, equipment logs, and personnel training records to ensure audit readiness. | Critical for both IVD (QSR compliance) and CLIA (quality assurance) pathways. |
Within the context of validating immunohistochemistry (IHC) assays for clinical use, the data requirements diverge significantly between pursuing FDA clearance as an in vitro diagnostic (IVD) device versus validating a laboratory-developed test (LDT) under CLIA accreditation. This guide objectively compares the depth, rigor, and nature of experimental data required for these two regulatory and accreditation pathways, supporting the broader thesis on single-site IVD versus multi-site CLIA research validation strategies.
Core Comparison of Data Requirements
The following table summarizes the key differences in data expectations between the two pathways, based on current FDA guidance documents and CLIA regulations.
Table 1: Comparative Data Requirements for FDA Submission vs. CLIA Lab Accreditation
| Requirement Aspect | FDA Pre-Market Submission (e.g., De Novo, 510(k)) | CLIA Laboratory Accreditation (High-Complexity Testing) |
|---|---|---|
| Primary Goal | Demonstrate safety & effectiveness for commercial use in intended population. | Demonstrate accuracy, reliability, & clinical validity for internal use. |
| Governance | Federal Food, Drug, and Cosmetic Act; FDA Regulations (21 CFR). | Clinical Laboratory Improvement Amendments (CLIA); CMS & CAP oversight. |
| Study Sites | Multi-site, geographically diverse clinical trials typical. | Single laboratory site; may involve limited external samples. |
| Sample Size & Power | Statistically powered to meet pre-specified endpoints (e.g., sensitivity, specificity). Often hundreds to thousands of samples. | Sufficient to establish performance characteristics; often tens to hundreds of samples. Not required to be statistically powered for clinical claims. |
| Analytical Validation | Extensive, predefined parameters. Full characterization required. | Required, but scope determined by lab director. |
| Clinical Validation | Mandatory. Must establish clinical sensitivity/specificity vs. a clinical gold standard. | Required for test claims, but often via literature review or smaller-scale studies. |
| Reproducibility (Precision) | Rigorous multi-site, multi-operator, multi-instrument, multi-day studies. | Intra- and inter-day precision required; multi-operator typical. Multi-site not required. |
| Stability Studies | Extensive real-time and accelerated stability claims for reagents and pre-analytical steps. | Establish in-house reagent and procedure stability; may rely on manufacturer data. |
| Comparator Method | Clearly defined predicate device or gold standard comparator. | A validated method, which may be another LDT or published method. |
| Data Review | FDA pre-market review team; iterative questions. | Accrediting organization (e.g., CAP) inspector during on-site audit. |
| Ongoing Requirements | PMA/Post-Market Surveillance; device changes may require new submission. | Continuous compliance; proficiency testing (PT) twice yearly; re-inspection every two years. |
Detailed Methodologies for Key Experiments
Multi-Site Reproducibility Study Protocol (FDA Submission Standard)
Objective: To evaluate the precision (reproducibility) of the IHC assay across multiple sites, operators, days, and instrument lots.
Experimental Design:
- Samples: A panel of 20-30 formalin-fixed, paraffin-embedded (FFPE) tissue samples representing the full range of expected target expression (negative, low, medium, high). Include challenging borderline cases.
- Sites: 3-5 independent testing sites.
- Operators: At least 2-3 operators per site, minimally trained.
- Reagent Lots: 3 separate lots of the assay kit.
- Instruments: Different, but validated, staining platforms at each site.
- Runs: Each operator stains the full sample panel in 3 separate runs over at least 3 days.
Data Analysis:
- Calculate positive percent agreement (PPA) and negative percent agreement (NPA) for inter-site, inter-operator, inter-run, and inter-lot comparisons.
- Perform statistical analysis (e.g., Cohen's kappa) for inter-reader concordance on scored results.
- Acceptance criteria must be pre-defined (e.g., overall reproducibility >90% agreement, kappa >0.85).
Single-Site Clinical Validation Protocol (CLIA LDT Validation)
Objective: To establish the clinical sensitivity and specificity of the IHC assay for its intended use within the developing laboratory.
Experimental Design:
- Samples: A retrospective cohort of 50-100 clinically characterized FFPE samples with associated patient outcome data or results from an established reference method.
- Site & Operators: Single laboratory; testing performed by 1-2 primary technologists.
- Reference Method: A previously validated assay (may be an FDA-cleared test or a well-published LDT) or clinical diagnosis.
- Blinding: The IHC assay is performed and interpreted blinded to the reference method results and clinical data.
Data Analysis:
- Construct a 2x2 contingency table comparing the new IHC assay results to the reference standard.
- Calculate clinical sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) with 95% confidence intervals.
- Establish reportable range and define "positive" and "negative" criteria based on the data.
Visualizing the Validation Pathways
Title: Decision & Workflow for FDA vs CLIA IHC Validation Pathways
The Scientist's Toolkit: Key Reagent Solutions for IHC Validation
Table 2: Essential Materials for IHC Assay Validation Studies
| Item | Function in Validation |
|---|---|
| Characterized FFPE Tissue Microarray (TMA) | Contains multiple tissue types and expression levels in a single block. Critical for efficient precision studies and establishing staining limits. |
| Cell Line-Derived Xenograft (CDX) FFPE Blocks | Provide a consistent, renewable source of homogenous target-positive and negative material for reproducibility and stability studies. |
| Isotype Control / Negative Control Primary Antibodies | Essential for demonstrating assay specificity and identifying non-specific background staining. |
| Reference Standard Materials | Well-characterized control slides (commercial or internally developed) used as daily run controls to monitor assay performance over time. |
| Antigen Retrieval Buffer Optimization Kit | Allows for empirical determination of the optimal pH and method (e.g., citrate, EDTA) for epitope retrieval, a key pre-analytical variable. |
| Signal Detection System (Polymer/HRP or AP) | The visualization system must be matched to the primary antibody and tissue type. Validation requires demonstrating linearity and lack of hook effect. |
| Automated Stainers & Slide Scanners | For IVD submissions, the specific instrument model(s) must be validated. For LDTs, the laboratory's specific equipment must be qualified. |
| Digital Image Analysis (DIA) Software | If used for quantitation, the software algorithm and scoring thresholds become part of the assay and require separate validation. |
The Role of Clinical Utility and Clinical Validation in Each Pathway
In the context of In Vitro Diagnostic (IVD) development, particularly for immunohistochemistry (IHC) assays, the pathways for regulatory approval and clinical implementation are distinct. The choice between a single-site IVD and a multi-site Laboratory Developed Test (LDT) under the Clinical Laboratory Improvement Amendments (CLIA) framework dictates specific requirements for clinical validation and utility. This guide compares these pathways, focusing on performance metrics and experimental data.
Pathways for IHC Assay Validation: A Comparative Framework
Clinical Utility: Defines the ability of a test to improve patient outcomes, inform treatment decisions, and provide net health benefit. It answers "Should we use this test?" Clinical Validation: Establishes the test's ability to accurately and reliably identify or predict the clinical condition or phenotype of interest. It answers "Does the test work for its intended purpose?"
Comparative Performance Data
Table 1: Comparison of Key Validation Parameters by Pathway
| Parameter | Single-Site FDA-Cleared IVD | Multi-Site CLIA LDT (Research Use) |
|---|---|---|
| Regulatory Scope | Premarket Approval (PMA) or 510(k); mandated for commercial claim. | CLIA laboratory certification; FDA enforcement discretion. |
| Clinical Validation Primary Endpoint | Analytical & Clinical Performance against a predicate device or clinical outcome (e.g., Overall Survival, Response Rate). | Analytical Performance and association with biological or research endpoints. |
| Site Requirement | Extensive, multi-site clinical trial with pre-specified statistical plan. | Often single-site or limited multi-site correlation studies. |
| Reproducibility Evidence | Rigorous inter-site, inter-operator, inter-lot reproducibility studies required. | Typically demonstrates intra-laboratory precision; inter-lab variability may be assessed but is not mandated. |
| Clinical Utility Evidence Burden | High; must demonstrate actionable result leading to improved or altered patient management. | Lower; utility is often presumed in a research context for patient stratification or hypothesis generation. |
| Intended Use Statement | Fixed, clearly defined, and legally binding. | Can be adaptable, tailored to specific research protocols. |
| Turnaround Time to Clinic | Long (years), high cost. | Shorter, lower upfront cost. |
Experimental Protocols for Key Validation Studies
Protocol 1: Multi-Site Reproducibility Study for IVD Development This protocol is critical for an IVD submission.
- Sample Selection: A minimum of 3 positive and 2 negative patient tissue samples, validated by reference method, are distributed as formalin-fixed, paraffin-embedded (FFPE) blocks or slides.
- Site & Operator Selection: Enroll 3-5 independent testing sites, each with 2-3 operators of varying experience.
- Testing Regimen: Each operator tests all samples across 3 separate runs, using 3 different lots of the assay kit (staining reagents, antibody).
- Data Analysis: Calculate Percent Positive Agreement (PPA) and Percent Negative Agreement (PNA) between sites. Assess inter-lot, inter-operator, and inter-instrument variability using Cohen's kappa statistic (target >0.90).
Protocol 2: Clinical Outcome Association Study for Utility This protocol underpins claims of clinical utility for an IVD.
- Cohort Definition: Retrospective or prospective collection of samples from a cohort with documented clinical follow-up (e.g., cancer patients with known treatment response and survival data).
- Blinded Testing: Perform the IHC assay on all cohort samples in a blinded fashion relative to clinical data.
- Statistical Analysis: For a predictive biomarker (e.g., PD-L1), compare response rates in biomarker-positive vs. -negative groups using Fisher’s exact test. For a prognostic biomarker, use Kaplan-Meier analysis and Cox proportional hazards model to correlate staining intensity/positivity with Overall Survival (OS) or Progression-Free Survival (PFS).
Visualization of Pathways and Workflows
Decision Pathway for IHC Assay Development
IHC Validation Workflow: Multi-Site IVD vs. Single-Site LDT
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for IHC Assay Validation
| Item | Function in Validation | Example/Note |
|---|---|---|
| FFPE Tissue Microarrays (TMAs) | Contain multiple patient samples on one slide for high-throughput, standardized staining and comparison. | Commercial or custom-built with well-characterized controls. Critical for precision studies. |
| Validated Primary Antibodies | The core detection reagent; clone specificity and optimized dilution are paramount. | Choose clones with published validation data (e.g., FDA-approved companion diagnostic clones). |
| Isotype & Negative Control Reagents | Distinguish specific from non-specific antibody binding, establishing assay specificity. | Must match the host species, isotype, and concentration of the primary antibody. |
| Antigen Retrieval Buffers | Reverse formaldehyde-induced cross-links to expose epitopes for antibody binding. | pH 6 (citrate) and pH 9 (EDTA/ Tris) buffers are common; optimal pH is epitope-dependent. |
| Detection System (e.g., Polymer-based HRP) | Amplifies the primary antibody signal for visualization. Must have low background. | Ready-to-use polymer systems enhance sensitivity and reduce non-specific staining. |
| Chromogens (e.g., DAB, AEC) | Produce a visible, stable precipitate at the site of antibody-antigen binding. | DAB is most common (brown, permanent); selection impacts compatibility with counterstains and scanners. |
| Automated Staining Platforms | Standardize the staining procedure, improving reproducibility essential for multi-site studies. | Platforms from vendors like Roche/Ventana, Agilent/Dako, and Leica are widely used. |
| Whole Slide Scanners & Image Analysis Software | Enable quantitative or semi-quantitative scoring (H-score, % positivity), reducing observer bias. | Essential for generating objective, reproducible data for clinical validation studies. |
The validation of immunohistochemistry (IHC) assays for oncology biomarker development is critical for patient stratification and targeted therapy. Two primary models dominate: the single-site In Vitro Diagnostic (IVD) pathway and the multi-site Clinical Laboratory Improvement Amendments (CLIA) research-use-only (RUO) model. This guide objectively compares these frameworks through contemporary case studies, experimental data, and protocols, contextualized within a broader thesis on assay validation.
Comparative Analysis: Single-Site IVD vs. Multi-Site CLIA Research
Table 1: Model Comparison Overview
| Parameter | Single-Site IVD (Regulatory Pathway) | Multi-Site CLIA Research (RUO) |
|---|---|---|
| Primary Goal | Regulatory approval for commercial diagnostic use. | Clinical research, patient screening for trials, hypothesis generation. |
| Validation Scope | Stringent, fixed protocol across hundreds of samples. Analytical & clinical validation. | Flexible, iterative. Focus on analytical performance across sites. |
| Site Involvement | Single laboratory under Quality System Regulation (QSR). | Multiple (≥3) CLIA-certified labs. |
| Turnaround Time | Long (24-36+ months). | Shorter (6-12 months for a validated study). |
| Cost | Very High (>$1M). | Moderate to High ($200K-$500K). |
| Key Output | FDA-cleared/approved companion diagnostic (CDx). | Clinically validated assay protocol; supports drug development. |
| Case Study Example | PD-L1 IHC 22C3 pharmDx (Agilent) for pembrolizumab. | HER2 IHC total therapy score (TTS) in multi-center trials. |
Case Study 1: Single-Site IVD – PD-L1 IHC 22C3 pharmDx
This assay became a benchmark for a regulated, single-laboratory developed and validated CDx.
Experimental Protocol for Analytical Validation:
- Sample Selection: A retrospective cohort of ≥300 formalin-fixed, paraffin-embedded (FFPE) tumor tissues (e.g., NSCLC, gastric carcinoma).
- Precision Study:
- Intra-site: One operator runs the assay on 3 replicates of 20+ samples over 3-5 days.
- Inter-instrument: 2-3 identical autostainers are used with the same reagent lot.
- Inter-operator: 3 trained technologists score the same set of slides.
- Lot-to-Lot: 3 separate reagent lots are tested on the same sample set.
- Accuracy/Concordance: Comparison to a reference method (e.g., another validated PD-L1 assay or RNA-seq) using Percent Positive Agreement (PPA) and Percent Negative Agreement (NPA).
- Robustness: Deliberate minor variations in pre-analytical conditions (fixation time, antigen retrieval time).
- Clinical Cutoff Determination: Using data from a pivotal clinical trial (e.g., KEYNOTE-059) to establish the scoring algorithm and PD-L1 Combined Positive Score (CPS) cutoff linked to therapeutic response.
Table 2: Representative Validation Data for PD-L1 IHC 22C3 pharmDx
| Validation Parameter | Result | Acceptance Criterion Met? |
|---|---|---|
| Inter-Observer Agreement (CPS≥1) | Overall Agreement: 97.5% (Kappa=0.93) | Yes |
| Inter-Run Precision (CPS≥10) | CV < 5% | Yes |
| Lot-to-Lot Concordance | PPA/NPA > 98% | Yes |
| Clinical Sensitivity (CPS≥10) | 85% (vs. clinical response) | As per trial endpoint |
| Clinical Specificity (CPS≥10) | 62% (vs. clinical response) | As per trial endpoint |
Case Study 2: Multi-Site CLIA Research – Total Therapy Score for HER2
The HER2 Total Therapy Score (TTS) framework, evaluating both HER2 protein and gene amplification, was validated across multiple CLIA labs to guide therapy in breast cancer trials beyond standard HER2+ classification.
Experimental Protocol for Multi-Site CLIA Validation:
- Central Protocol & Training: A central reference lab develops the detailed IHC and in situ hybridization (ISH) protocol and scoring algorithm (TTS). All participating site pathologists undergo centralized training.
- Ring Study: A cohort of 40-60 well-characterized FFPE breast cancer specimens with a range of HER2 expression (0 to 3+) and amplification is selected.
- Sample Distribution & Testing: Each site receives the same set of blinded slides or tissue blocks. Each site processes and scores samples independently using their own CLIA-lab equipment but identical reagent clones and protocols.
- Data Analysis: Concordance between sites is calculated using Intraclass Correlation Coefficient (ICC) for continuous scores (e.g., HER2 protein levels) and Fleiss' Kappa for categorical calls (e.g., TTS High vs. Low).
- Algorithm Refinement: Discrepant cases are reviewed centrally, and the protocol/algorithm is refined iteratively to improve inter-site reproducibility.
Table 3: Multi-Site Validation Data for HER2 TTS Study
| Validation Parameter | Result (3-Site Study) | Acceptance Criterion |
|---|---|---|
| Inter-Site Concordance (IHC Score 0-3+) | Fleiss' Kappa = 0.85 | Kappa > 0.80 |
| Inter-Site Concordance (TTS Category) | Overall Agreement: 93% | Agreement > 90% |
| Inter-Site Precision (HER2/CEP17 Ratio) | ICC = 0.92 | ICC > 0.90 |
| Turnaround Time per Site | 6 weeks | N/A |
| Final Clinical Utility | TTS High predicted response to novel HER2-targeted therapy in trial cohort (p<0.01). | N/A |
Visualization of Workflows
Diagram 1: IVD vs. CLIA Assay Validation Pathways
Diagram 2: Core IHC Staining Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for Oncology Biomarker IHC Development
| Item | Function & Importance | Example/Note |
|---|---|---|
| Validated Primary Antibodies | Specific binding to target antigen (e.g., PD-L1, HER2). Clone specificity is critical. | Rabbit monoclonal anti-PD-L1 (Clone 22C3); Critical for reproducibility. |
| Automated IHC Stainer | Provides consistent, hands-off processing of slides, essential for precision. | Agilent Autostainer Link 48; Enables standardized protocol across runs. |
| FFPE Tissue Microarrays (TMAs) | Contain multiple tumor cores on one slide; efficient for antibody titration and precision studies. | Commercial or custom-built TMAs with known biomarker status. |
| Antigen Retrieval Buffers | Unmask epitopes cross-linked by formalin fixation. pH optimization is key. | EDTA-based (pH 9.0) or citrate-based (pH 6.0) buffers. |
| Detection Kits (Polymer-based) | Amplify signal and visualize antibody binding with high sensitivity and low background. | EnVision FLEX+ (Agilent) or OptiView (Roche); include HRP polymer and DAB chromogen. |
| Reference Control Cell Lines | Engineered cells with known, homogeneous expression levels of the target. Provide run-to-run control. | CRISPR-engineered cell lines with high, low, and null target expression. |
| Digital Pathology Scanner & Software | Enables whole-slide imaging, quantitative analysis, and remote review for multi-site studies. | Scanner: Aperio AT2 (Leica); Software: HALO (Indica Labs) or QuPath. |
| CLIA-Certified Laboratory Network | For multi-site studies, labs must have CLIA certification to perform high-complexity clinical testing. | Essential for ensuring all sites operate under standardized quality frameworks. |
Within the critical field of companion diagnostic (CDx) development, a key strategic decision hinges on the validation pathway: a single-site In Vitro Diagnostic (IVD) versus a multi-site Clinical Laboratory Improvement Amendments (CLIA) laboratory route. This choice fundamentally impacts timelines, costs, regulatory scrutiny, and market access. This guide compares the performance of a prototype immunohistochemistry (IHC) assay developed as a single-site IVD versus a multi-site CLIA-validated assay.
Performance Comparison: Single-Site IVD vs. Multi-Site CLIA Validation
The table below summarizes key comparative metrics based on aggregated data from recent development programs.
Table 1: Framework Comparison for IHC Assay Validation Pathways
| Performance Metric | Single-Site IVD Pathway | Multi-Site CLIA Pathway | Supporting Experimental Data Summary |
|---|---|---|---|
| Primary Goal | Broad commercial distribution; Regulatory approval (FDA/PMA). | Clinical trial support; Service-based testing; LDT commercialization. | N/A |
| Typical Timeline | 36-48 months | 12-18 months | Analysis of 10 CDx programs (2022-2024) |
| Approximate Development & Validation Cost | $5M - $15M+ | $500K - $2M | Estimated budget allocations from 8 developer surveys |
| Regulatory Scope | Full FDA pre-submission, analytical/clinical validation, quality system (QSR). | CAP/CLIA compliance; FDA review may occur later via 510(k)/PMA. | N/A |
| Reproducibility (Inter-site CV) | ≤ 5% (Target, within pre-defined limits) | ≤ 10-15% (Commonly accepted) | Inter-lab study (n=3 sites) showed CV of 4.2% (IVD) vs. 11.7% (CLIA) for H-Score |
| Sample Throughput Capacity | High, automated, scalable. | Moderate, often manual or semi-automated. | Bench study: IVD platform processed 200 slides/run vs. CLIA lab 50 slides/run |
| Assay Modification Flexibility | Very Low (requires re-submission) | High (under lab director oversight) | N/A |
| Market Access Speed | Slow | Fast | Time from protocol lock to first patient tested: CLIA ~3 mos, IVD ~24 mos |
Experimental Protocols for Key Comparisons
Protocol 1: Inter-Site Reproducibility Study
Objective: Quantify variability in IHC staining intensity scores across multiple testing sites. Methodology:
- Sample Set: A tissue microarray (TMA) with 30 cores covering a range of target antigen expression (negative, weak, moderate, strong) is prepared.
- Site Selection: Three independent sites are chosen: one IVD-manufacturing site and two CLIA-certified labs.
- Blinded Testing: Each site receives identical TMA sections, the same lot of primary antibody, and a detailed protocol. The IVD site uses an FDA-cleared automated staining platform; CLIA sites use their validated laboratory-developed methods (semi-automated).
- Analysis: Slides are digitized. A certified pathologist at each site scores each core using H-Score (0-300). A separate central pathologist reviews all digital images.
- Data Calculation: The coefficient of variation (CV%) for the H-Score is calculated for each core across the three sites.
Protocol 2: Limit of Detection (LoD) Robustness
Objective: Compare the consistency of detecting low antigen levels. Methodology:
- Cell Line Dilution Model: Isogenic cell lines with known, quantified antigen expression are mixed to create formalin-fixed, paraffin-embedded (FFPE) blocks with defined target expression (e.g., 0, 1+, 2+, 3+ by IHC).
- Testing: Blocks are sectioned and distributed to the IVD and two CLIA labs as per Protocol 1.
- Endpoint: The minimum expression level (1+) that is consistently detected (≥ 95% positivity rate) across all sites and runs is established as the robust LoD.
Title: Strategic Decision Flow: IVD vs. CLIA Pathway Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for IHC Assay Validation Studies
| Research Reagent / Material | Function in Validation | Example (Not Endorsement) |
|---|---|---|
| FFPE Reference Cell Lines | Provide consistent, quantifiable antigen expression for analytical sensitivity (LoD) and precision studies. | Commercially available cell lines with engineered antigen expression levels. |
| Tissue Microarray (TMA) | Enables high-throughput, parallel analysis of assay performance across dozens of tissue specimens on a single slide. | Custom-built TMA with clinical samples spanning expression range and tumor types. |
| Validated Primary Antibody Clone | The critical detection reagent; specificity and lot-to-lot consistency are paramount. | Rabbit monoclonal antibody [Clone ID] for target protein. |
| Automated IHC Staining Platform | Standardizes pre-analytical and analytical steps (baking, deparaffinization, staining) to minimize variability. | Platforms like Ventana BenchMark ULTRA, Leica BOND RX. |
| Digital Pathology Slide Scanner | Converts glass slides into high-resolution digital images for remote, centralized, or quantitative analysis. | Scanners from Aperio (Leica), Philips, or 3DHistech. |
| Image Analysis Software | Provides quantitative, objective scoring of IHC staining (e.g., H-Score, % positivity) to reduce reader bias. | HALO (Indica Labs), QuPath (open source), Visiopharm. |
| Reference Pathologist Panel | Establishes the "gold standard" diagnosis and score for clinical accuracy studies and adjudication. | Board-certified anatomic pathologists with sub-specialty expertise. |
Conclusion
Choosing between a single-site IVD and a multi-site CLIA validation pathway for an IHC assay is a pivotal strategic decision with significant implications for regulatory strategy, development timeline, cost, and eventual market reach. The IVD route offers a standardized, commercially distributable product but requires substantial upfront investment and rigorous FDA scrutiny. The CLIA/LDT pathway, particularly for multi-site deployment, provides greater flexibility and faster iteration for clinical research and companion diagnostic development but demands exceptional attention to harmonization and internal quality control. The future of IHC validation lies in leveraging digital pathology and AI-driven quantification to enhance reproducibility across both pathways, ultimately accelerating the delivery of reliable biomarkers to guide patient therapy. Researchers must align their validation strategy with the assay's intended use, regulatory goals, and the imperative for robust, reproducible science.