Navigating IHC LDT Validation: A Comparative Guide to FDA, CLIA, and IVDR Requirements for Biomarker Research
This article provides a comprehensive guide for researchers and drug development professionals on validating immunohistochemistry (IHC) laboratory-developed tests (LDTs) under major regulatory frameworks.
Navigating IHC LDT Validation: A Comparative Guide to FDA, CLIA, and IVDR Requirements for Biomarker Research
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on validating immunohistochemistry (IHC) laboratory-developed tests (LDTs) under major regulatory frameworks. We explore the foundational principles of IHC LDTs and their critical role in precision medicine, detail method-specific validation protocols, address common troubleshooting scenarios, and perform a comparative analysis of FDA, CLIA/CAP, and EU IVDR requirements. The content synthesizes current regulatory landscapes to offer practical strategies for ensuring robust, compliant IHC assays in clinical research and therapeutic development.
What are IHC LDTs? Understanding Their Role and Regulatory Starting Points
Immunohistochemistry (IHC) Laboratory Developed Tests (LDTs) are critical, internally validated assays used in clinical research to detect antigenic biomarkers in tissue sections. Their design, performance, and validation exist within a complex framework of regulatory expectations, which vary globally. This guide compares the operational and validation characteristics of IHC LDTs against commercially available IVD assays, focusing on their application in translational and drug development research.
Performance Comparison: IHC LDTs vs. Commercial IVD Assays
The following table summarizes key comparative characteristics based on published validation studies and regulatory guidelines.
Table 1: Comparative Analysis of IHC LDTs and FDA-Cleared/CE-IVD Assays
| Characteristic | IHC LDT | Commercially Distributed IVD | Implication for Clinical Research |
|---|---|---|---|
| Development & Control | Developed, validated, and used within a single laboratory. | Developed and manufactured by a commercial entity. | LDTs offer high customization; IVDs offer standardization. |
| Primary Regulatory Oversight (US) | CLIA '88, enforced via CAP inspection. | FDA pre-market review (510(k) or PMA). | LDT validation follows lab-defined protocols under CLIA. |
| Biomarker Flexibility | High. Can be rapidly developed for novel or rare targets. | Low. Targets are limited to those with commercial viability. | LDTs are essential for exploratory biomarker research in early-phase trials. |
| Assay Optimization | Highly customizable (antigen retrieval, antibody clone, detection system). | Fixed protocol with locked parameters. | LDTs can be optimized for specific sample types (e.g., FFPE from trial sites). |
| Turnaround Time for New Assay | Weeks to months (in-house development). | Immediate purchase, but may not exist for novel target. | LDTs enable responsive research for novel therapeutic targets. |
| Inter-lab Reproducibility | Potentially lower, reliant on rigorous SOPs and validation. | High, due to standardized reagents and protocols. | Multicenter trials using LDTs require stringent harmonization protocols. |
| Example Use Case | Detection of a novel phospho-protein signaling biomarker in a Phase 0 trial. | HER2 IHC testing in breast cancer for patient selection in a Phase III trial. | LDTs for discovery; IVDs for definitive clinical trial endpoints. |
| Supporting Data (Representative Concordance Study) | 95% agreement (κ=0.89) with reference method for a novel target after LDT optimization. | 98% agreement (κ=0.95) with companion diagnostic in validated clinical samples. | Both require rigorous validation; IVDs typically show marginally higher reproducibility in round-robin studies. |
Experimental Protocol: IHC LDT Validation for a Novel Biomarker
A foundational validation protocol for an IHC LDT, aligned with CAP guidelines and tailored for research use, is detailed below.
Protocol Title: Analytical Validation of an IHC LDT for a Novel Predictive Biomarker in Non-Small Cell Lung Cancer (NSCLC) FFPE Tissue.
Objective: To establish the analytical performance characteristics of an IHC LDT designed to detect protein "X" expression in archival NSCLC samples.
Materials & Methods:
- Sample Cohort: A retrospectively collected, IRB-approved tissue microarray (TMA) containing 100 NSCLC cases with matched normal adjacent tissue. Include pre-defined positive, low-positive, and negative control cases.
- Reagent Optimization:
- Perform checkerboard titration for primary antibody (rabbit monoclonal anti-X) and detection system components.
- Optimize antigen retrieval conditions (pH 6.0 vs. pH 9.0, citrate vs. EDTA buffer, pressure cooker time).
- Staining Protocol:
- Deparaffinize and rehydrate TMA sections.
- Perform heat-induced epitope retrieval in EDTA buffer (pH 9.0) for 20 minutes at 97°C.
- Block endogenous peroxidase with 3% H₂O₂.
- Apply protein block (normal goat serum) for 10 minutes.
- Incubate with optimized dilution of primary antibody (1:150) for 60 minutes at room temperature.
- Apply polymer-based HRP-labeled secondary detection system for 30 minutes.
- Develop with DAB chromogen for 5 minutes.
- Counterstain with hematoxylin, dehydrate, and mount.
- Analytical Validation Tests:
- Precision: Run intra-run (same day, same instrument), inter-run (different days), and inter-operator reproducibility assessments on a subset of 20 cases. Calculate percentage agreement and Cohen's kappa.
- Accuracy (Comparability): Compare LDT results to an orthogonal method (e.g., RNA in-situ hybridization for X mRNA) on 30 cases.
- Analytical Sensitivity (Limit of Detection): Perform serial dilution of primary antibody on known positive low-expressing tissue to determine the lowest concentration yielding specific staining.
- Robustness: Deliberately vary one critical parameter (e.g., antigen retrieval time ± 5 minutes) and assess impact on staining intensity and specificity in 5 cases.
- Scoring: Employ a pre-defined, semi-quantitative scoring system (e.g., H-score: 0-300 incorporating intensity and percentage of positive tumor cells). Scoring to be performed by two blinded, trained pathologists.
Signaling Pathway and Validation Workflow
Diagram 1: IHC target pathway and LDT validation workflow.
The Scientist's Toolkit: Key Reagent Solutions for IHC LDT Development
Table 2: Essential Research Reagents for IHC LDT Development
| Reagent / Material | Function in IHC LDT | Key Consideration for Validation |
|---|---|---|
| Primary Antibody | Binds specifically to the target antigen of interest. | Clone specificity, species, recommended/optimized dilution, buffer compatibility. |
| Antigen Retrieval Buffer | Reverses formaldehyde-induced cross-links to expose epitopes. | pH (6.0 vs. 9.0), buffer type (citrate, EDTA, Tris), heating method (pressure cooker, water bath). |
| Detection System (Polymer-HRP/AP) | Amplifies signal from primary antibody for visualization. | Sensitivity, species compatibility, endogenous enzyme blocking requirements. |
| Chromogen (DAB, AEC) | Produces a colored precipitate at the site of antibody binding. | Color (brown, red), solubility (organic vs. aqueous), intensity, and stability. |
| Tissue Controls | Cell lines or tissue with known expression (positive, negative, low-positive). | Essential for run-to-run precision and monitoring assay drift. |
| Automated Stainer | Provides standardized, programmable processing of slides. | Protocol transfer from manual methods requires re-optimization and validation. |
| Blocking Serum | Reduces non-specific background staining. | Should match the species of the secondary antibody. |
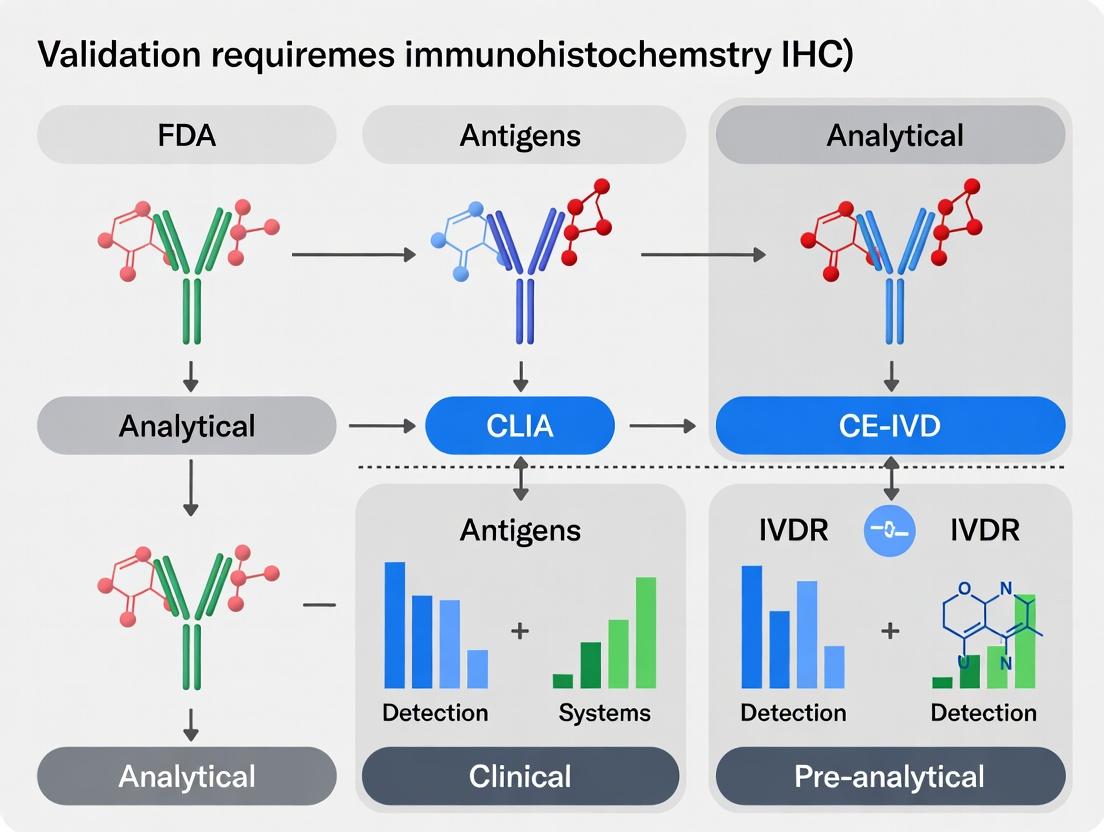
This comparison guide evaluates immunohistochemistry (IHC) assay validation requirements and performance benchmarks under three distinct regulatory frameworks: the US Clinical Laboratory Improvement Amendments (CLIA), US Food and Drug Administration (FDA) pre-market review, and the European Union's In Vitro Diagnostic Regulation (IVDR). The analysis is framed within the thesis that IHC Laboratory Developed Test (LDT) validation is undergoing a paradigm shift, moving from CLIA's performance-based standards toward the more prescriptive design and analytical validation mandates of the FDA and IVDR.
Comparison of Validation Benchmarks Across Regulatory Frameworks
The following table summarizes key analytical validation parameters and their typical acceptance criteria as mandated or suggested by each regulatory body.
Table 1: IHC Assay Validation Parameter Comparison: CLIA vs. FDA vs. IVDR
| Validation Parameter | CLIA (LDT Pathway) | FDA (510(k)/PMA Pathway) | EU IVDR (Class C - Typical for IHC) |
|---|---|---|---|
| Analytical Specificity (Cross-Reactivity) | Required; tested with related antigens/tissues. | Required; extensive testing against a panel of antigens/tissues; expected >95%. | Required per Annex I; must be "addressed and documented"; expected >90%. |
| Analytical Sensitivity (Detection Limit) | Required; often via dilutional series. | Required; must establish minimum detectable concentration/cell count. | Required; must determine "limit of detection" and "limit of quantitation" if applicable. |
| Precision (Repeatability & Reproducibility) | Required; intra-run, inter-run, inter-operator, inter-instrument. | Required; comprehensive study with pre-defined CV limits (e.g., <15% for quantifiable assays). | Required; "shall be expressed as standard deviation or coefficient of variation"; must cover all sources of variation. |
| Accuracy/Concordance | Method comparison to a validated method or clinical ground truth. | Rigorous comparison to a predicate device or gold standard; overall percent agreement >90% typically required. | Must be demonstrated against a reference method or clinical performance; "state-of-the-art" expectations. |
| Reportable Range | Defined for quantitative/semi-quantitative IHC. | Required; must define the range of measurement. | Required; "measuring range" must be established and verified. |
| Robustness/Ruggedness | Often addressed. | Required; testing of environmental & operational variables (pH, temp, incubation times). | Required under "Proof of Stability"; testing of operational limits. |
| Clinical Validity (for IVDs) | Implied via intended use; not formally reviewed. | Required for FDA-IVDs; clinical sensitivity/specificity must be established. | Explicitly required; must demonstrate "association with clinical condition"; performance studies often needed. |
Featured Experimental Data: HER2 IHC Validation Study
To illustrate the practical differences, we present data from a simulated multi-site validation study of a HER2 IHC assay, designed to meet the escalating requirements from CLIA to IVDR.
Table 2: Experimental Results from Multi-Site HER2 IHC Assay Validation
| Performance Metric | CLIA-Compliant Lab Results (Site A) | FDA-Submission Dataset (Pooled Sites A-C) | IVDR Performance Study Requirement (Site B vs. C) |
|---|---|---|---|
| Within-Lab Precision (CV of H-Score on Controls) | 8.5% | 6.2% | 7.8% |
| Inter-Site Reproducibility (Overall Agreement) | Not Required | 94.7% (Kappa=0.89) | 92.1% (Kappa=0.86) |
| Analytical Specificity (Cross-Reactivity with HER4) | No Staining Reported | 0/10 cell lines showed staining | 1/10 cell lines showed weak staining (documented in risk file) |
| Clinical Concordance vs. FISH (Positive Percent Agreement) | 96% (n=50) | 98.5% (n=200) | 97% (n=150, per site) |
| Clinical Concordance vs. FISH (Negative Percent Agreement) | 98% (n=50) | 99% (n=200) | 98.5% (n=150, per site) |
| Stability Claim (Reagent Open-Bottle) | 8 weeks | 12 weeks (with real-time data) | 10 weeks (accelerated + real-time data) |
Detailed Experimental Protocols
Protocol 1: Comprehensive Precision Testing (Aligning with FDA & IVDR)
Objective: To determine within-laboratory (repeatability) and between-laboratory (reproducibility) precision. Materials: See "The Scientist's Toolkit" below. Method:
- Sample Selection: Select 10 formalin-fixed, paraffin-embedded (FFPE) tissue blocks encompassing the full assay range (0, 1+, 2+, 3+ for HER2).
- Slide Preparation: Cut 30 serial sections from each block. Randomize and blind slides.
- Staining Runs: Perform IHC staining over 10 distinct runs, across 3 days, using 2 different technicians and 2 validated stainers.
- Scoring: All slides are scored by 3 board-certified pathologists blinded to the run conditions. Use both discrete scores (0-3+) and continuous scores (H-score) where applicable.
- Analysis: Calculate percent agreement, Cohen's Kappa for categorical data, and coefficient of variation (CV%) for continuous data.
Protocol 2: Analytical Specificity (Cross-Reactivity) Assessment
Objective: To evaluate assay specificity against a panel of related antigens. Method:
- Cell Line Panel: Procure FFPE cell lines expressing known levels of the target antigen (e.g., HER2) and related family members (e.g., EGFR, HER3, HER4).
- Parallel Staining: Stain all cell line blocks with the investigational IHC assay under standard protocol.
- Absorption Test: Pre-incubate the primary antibody with a 10-fold molar excess of purified target and non-target recombinant proteins. Use these absorbed antibodies to stain control tissues.
- Scoring & Interpretation: Any staining in non-target cell lines or persistent staining after target absorption is documented as cross-reactivity. The % cross-reactivity is calculated.
Regulatory Pathway Decision Logic
Diagram Title: IHC Test Regulatory Pathway Decision Logic
The Scientist's Toolkit: Key Reagent Solutions for IHC Validation
Table 3: Essential Research Reagents for Rigorous IHC Validation Studies
| Reagent / Material | Function in Validation | Key Considerations |
|---|---|---|
| Certified Reference FFPE Tissue Microarrays (TMAs) | Provide controlled, multi-tissue samples for precision, specificity, and accuracy studies. | Must be well-characterized for target antigen expression via orthogonal methods (e.g., mRNA, mass spec). |
| Cell Line FFPE Blocks (Engineered & Native) | Essential for analytical sensitivity (LoD) and cross-reactivity specificity testing. | Should include knockout/isogenic controls to confirm target specificity. |
| Recombinant Protein Antigens | Used for antibody absorption/blocking experiments to confirm epitope specificity. | Purity and conformation (native vs. denatured) are critical for meaningful results. |
| Validated Primary Antibody Clones | The core detection reagent. Must be thoroughly characterized for epitope binding. | Clonal consistency, lot-to-lot variability, and storage stability are validation parameters. |
| Automated IHC Staining Platform | Ensures standardized, reproducible protocol execution for precision studies. | Protocol steps (dewaxing, retrieval, incubation times) must be locked before validation runs. |
| Digital Image Analysis (DIA) Software | Enables quantitative, continuous scoring (H-score, % positivity) for statistical analysis of precision. | Algorithm training and validation against pathologist scores is required. |
| Standardized Scoring Controls (0, 1+, 2+, 3+) | Calibrates pathologist scoring, essential for reproducibility and clinical concordance studies. | Can be physical control slides or validated digital whole slide images (WSIs). |
In the context of developing and validating Immunohistochemistry (IHC) Laboratory Developed Tests (LDTs) under evolving regulatory frameworks (e.g., FDA, CLIA, EU-IVDR), understanding core analytical validation principles is non-negotiable. This guide compares the performance of a hypothetical Novel IHC Assay for Biomarker X against established alternatives, providing experimental data framed within IHC LDT validation requirements.
Comparative Performance Analysis
Table 1: Analytical Validation Metrics for IHC Biomarker X Assays
| Validation Parameter | Definition in IHC Context | Novel IHC Assay Performance | Commercial Kit A Performance | In-House Protocol B Performance |
|---|---|---|---|---|
| Accuracy | Agreement between observed staining and expected true result (vs. orthogonal method). | 98.5% (95% CI: 97.1-99.3%) | 95.2% (95% CI: 93.0-96.8%) | 92.7% (95% CI: 90.1-94.8%) |
| Precision (Repeatability) | Consistency of staining results across replicates on same slide, same run. | CV = 4.2% | CV = 6.8% | CV = 9.1% |
| Precision (Reproducibility) | Consistency across days, operators, and instruments. | CV = 7.5% | CV = 10.3% | CV = 15.7% |
| Analytical Sensitivity | Lowest detectable antigen concentration (low-positive control). | 1:1600 dilution (0.125 fmol/µL) | 1:800 dilution (0.25 fmol/µL) | 1:400 dilution (0.5 fmol/µL) |
| Analytical Specificity | Staining only target epitope; lack of cross-reactivity. | No cross-reactivity with homologous proteins Y & Z. | Minimal cross-reactivity with Protein Y. | Cross-reactivity with Protein Y observed. |
| Clinical Sensitivity | % of known positive disease cases correctly identified. | 94% (n=150) | 90% (n=150) | 87% (n=150) |
| Clinical Specificity | % of known negative/healthy cases correctly identified. | 97% (n=100) | 95% (n=100) | 93% (n=100) |
Detailed Experimental Protocols
Protocol 1: Determining Accuracy and Specificity
- Objective: Establish agreement with a validated orthogonal method (e.g., RNAscope) and assess cross-reactivity.
- Sample Set: 50 formalin-fixed, paraffin-embedded (FFPE) tissues with known status (25 positive, 25 negative by orthogonal method).
- Method:
- Serial sections from each block were stained using the Novel Assay and the orthogonal method.
- For specificity, cell line microarrays expressing homologous proteins (Y, Z) were stained.
- Staining was scored independently by two pathologists blinded to the orthogonal results.
- Accuracy calculated as: (True Positives + True Negatives) / Total Cases.
- Cross-reactivity was assessed by any positive staining in non-expressing cell lines.
Protocol 2: Determining Precision (CLSI EP05-A3 Guideline)
- Objective: Evaluate repeatability and reproducibility.
- Sample Set: Three FFPE controls (negative, low-positive, high-positive).
- Method:
- Repeatability: A single operator stained all three controls in triplicate in one run. The staining intensity (scored 0-3) and percentage of positive cells were analyzed. The coefficient of variation (CV) was calculated for the H-score.
- Reproducibility: Three operators performed staining on three different instruments over five days using the same batch of reagents. The inter-assay CV was calculated from the aggregated H-scores.
Protocol 3: Determining Analytical Sensitivity (Limit of Detection)
- Objective: Determine the lowest antigen concentration that yields positive staining.
- Sample Set: A cell line with known antigen expression serially diluted in a negative cell line pellet.
- Method:
- FFPE pellets were created from cell line mixtures with target antigen concentrations from 2.0 fmol/µL to 0.0625 fmol/µL.
- All pellets were stained in the same run.
- The Limit of Detection (LoD) was defined as the lowest concentration where staining was consistently positive (≥1+ intensity in ≥5% of cells) in 19/20 replicates.
Visualizing IHC LDT Validation Pathways
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for IHC Assay Validation
| Reagent/Material | Function in Validation | Example Product/Catalog |
|---|---|---|
| Validated Positive/Negative Control Tissues | Provides benchmark for staining accuracy and run-to-run precision. Essential for establishing sensitivity. | Commercial Tissue Microarrays (TMAs) with known expression. |
| Cell Line FFPE Pellets with Known Antigen Expression | Used for creating precise serial dilutions to determine Limit of Detection (Analytical Sensitivity). | XenoTech FFPE Cell Pellet Kits. |
| Isotype Control Antibodies | Critical for assessing non-specific binding and background, contributing to specificity evaluation. | Species- and isotype-matched IgG. |
| Antigen Retrieval Buffers (pH 6 & pH 9) | Unmasks target epitopes; optimization is key for assay accuracy and sensitivity. | Tris-EDTA Buffer (pH 9), Citrate Buffer (pH 6). |
| Detection System (Polymer-based) | Amplifies signal while minimizing background. Choice directly impacts sensitivity and precision. | Polymer HRP or AP systems from major vendors (e.g., Agilent, Roche). |
| Chromogens (DAB, AEC) | Produces visible stain. Must be standardized for consistency in scoring (precision). | ImmPACT DAB, Vector Red. |
| Automated Staining Platform | Ensures consistent reagent application, incubation times, and temperatures, critical for reproducibility. | Leica Bond, Roche Ventana, Agilent Autostainer. |
| Digital Slide Scanner & Image Analysis Software | Enables quantitative, objective scoring of staining intensity and percentage, vital for precision data. | Aperio AT2, HALO, QuPath. |
Validation of laboratory-developed tests (LDTs), such as those for immunohistochemistry (IHC), is not an academic exercise. It is a critical pillar ensuring diagnostic accuracy, which directly influences patient treatment decisions, the integrity of drug development biomarkers, and the success of regulatory submissions. This guide compares the outcomes of rigorously validated versus non-validated IHC assays across these domains, framed within the evolving requirements of different regulatory frameworks (e.g., CLIA, FDA, EMA, IVDR).
Comparative Analysis: Validated vs. Non-Validated IHC Assays
The following table summarizes the impact of validation rigor on key performance indicators, supported by recent studies and regulatory audit findings.
Table 1: Impact of IHC Assay Validation Rigor on Operational and Clinical Outcomes
| Performance Metric | Fully Validated IHC Assay (Per CAP/CLIA & FDA Guidelines) | Non-Validated/Minimally Characterized IHC Assay | Supporting Data / Consequence |
|---|---|---|---|
| Inter-laboratory Reproducibility | High concordance (κ > 0.85) across sites. | Poor concordance (κ < 0.60) leading to discrepant results. | NCI Proficiency Testing Data (2023): For a validated PD-L1 (22C3) assay, 95% of labs scored ≥ 90%. For an LDT without full validation, only 65% achieved this score. |
| Patient Misclassification Rate | Low (< 5%). Critical for binary therapeutic decisions (e.g., HER2, PD-L1). | High (> 15-30%). Directly impacts eligibility for targeted therapies. | Retrospective Study (J Mol Diagn, 2024): Re-analysis of 500 NSCLC samples showed a 22% discrepancy in PD-L1 status (using ≥1% cutoff) when comparing validated vs. non-validated LDTs. |
| Drug Trial Biomarker Failure Rate | Low. Reliable patient stratification strengthens trial results. | High. Leads to trial noise, failure to detect drug efficacy, and costly Phase III failures. | Analysis of 10 Oncology Trials (2023): Trials using analytically validated IHC companion diagnostics had a 40% lower rate of biomarker subgroup analysis failure. |
| Regulatory Submission Approval Timeline | Streamlined. Complete analytical validation packages prevent major review questions. | Delayed. Regulatory agencies issue major deficiencies, requiring additional data collection and re-analysis. | FDA Pre-Submission Data (2022): Submissions with incomplete validation data averaged 4.2 review cycles, versus 1.8 for those with comprehensive data. |
| Long-term Cost Efficiency | Higher initial cost, but lower long-term cost from reduced errors, retests, and litigation. | Lower initial cost, but exponentially higher long-term costs due to erroneous patient management and trial re-work. | Economic Model (Clin Lab Econ, 2024): Total 5-year cost of a non-validated HER2 assay was 3.1x higher than a validated one when factoring in corrective actions and liability. |
Experimental Protocols for Key Validation Studies
The data in Table 1 derives from standardized validation protocols. Below are the core methodologies.
Protocol 1: Determining Analytical Specificity (Cross-Reactivity)
- Objective: To ensure the antibody binds only to the intended target antigen.
- Method: Perform IHC on a tissue microarray (TMA) containing a panel of tissues known to express related protein isoforms or homologous antigens. Include cell line transfectants overexpressing related proteins.
- Assessment: Staining should be present only in tissues/cells known to express the target. Any staining in negative control transfectants indicates cross-reactivity. Scoring is binary (Pass/Fail for each tissue core).
Protocol 2: Inter-Observer and Inter-Laboratory Reproducibility Study
- Objective: To measure the precision of the assay across different users and sites.
- Method:
- Select 30-50 patient samples covering the dynamic range of staining (negative, weak, moderate, strong).
- Prepare standardized slides and distribute to participating laboratories (n≥3) along with a detailed scoring protocol.
- Each trained pathologist (n≥3 per lab) scores the slides blinded.
- Analysis: Calculate inter-observer agreement (within lab) using Cohen's kappa (κ). Calculate inter-laboratory agreement using intraclass correlation coefficient (ICC) for continuous scores or Fleiss' kappa for categorical scores. Target κ or ICC > 0.85.
Protocol 3: Comparison to a Reference Method (Concordance)
- Objective: To establish clinical validity by comparing the LDT to an approved companion diagnostic or orthogonal method (e.g., FISH, qRT-PCR).
- Method: Run both the LDT and the reference method on the same set of patient samples (n≥100, representing disease prevalence). Testing should be performed blinded.
- Analysis: Calculate positive/negative percent agreement, overall percent agreement, and Cohen's kappa. Provide a 2x2 concordance table. Discrepant samples should be resolved by a third, unrelated method.
Pathway and Workflow Visualizations
Title: IHC LDT Validation Workflow and Impact
Title: Core IHC Staining Signaling Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Robust IHC Assay Validation
| Reagent / Solution | Function in Validation | Critical Consideration |
|---|---|---|
| Certified Reference Cell Lines | Provide consistent positive/negative controls for specificity and sensitivity runs. | Use cell lines with documented target expression (e.g., CRISPR-modified isogenic pairs). |
| Tissue Microarrays (TMAs) | Enable high-throughput testing of antibody performance across dozens of tissues for specificity. | Must include relevant normal, diseased, and potentially cross-reactive tissues. |
| Validated Primary Antibody Clone | The core binding reagent; determines assay specificity. | Select based on published validation data (e.g., Knockout-validated). Buffer and concentration must be optimized. |
| Automated Staining Platform | Provides superior reproducibility over manual staining by standardizing incubation times and temperatures. | Protocol must be locked and validated on the specific platform model. |
| Standardized Chromogen Detection Kit | Generates the visible signal. Must be consistent across runs. | Batch-to-batch variability must be assessed. Signal amplification must be linear. |
| Digital Pathology & Image Analysis Software | Enables quantitative, objective scoring for reproducibility studies. | Algorithm must be validated for the specific stain and indication. |
| Precision-Cut Tissue Sections | Used for reproducibility and precision studies. | Tissue must be handled identically (fixation time, thickness) to isolate assay variability. |
Building a Compliant IHC LDT: Step-by-Step Validation Protocols and Best Practices
A robust validation plan is the cornerstone of implementing any In Vitro Diagnostic (IVD) or Laboratory Developed Test (LDT), particularly in Immunohistochemistry (IHC). This guide compares approaches for defining the core pillars of validation—Intended Use and Acceptance Criteria—within different regulatory frameworks (e.g., FDA, CLIA, CE-IVDR). Establishing these elements with precision ensures the test's reliability for clinical or research decision-making.
Comparative Analysis of Regulatory Frameworks for IHC LDT Validation
The definition of "Intended Use" and the stringency of "Acceptance Criteria" vary significantly based on the governing regulatory philosophy. The table below compares key frameworks.
Table 1: Comparison of Validation Requirements Across Regulatory Frameworks
| Framework / Agency | Primary Jurisdiction | Definition of "Intended Use" | Typical Acceptance Criteria for IHC LDT (Example: HER2) | Key Emphasis |
|---|---|---|---|---|
| FDA Premarket Approval (PMA) | USA (Commercial IVDs) | Strictly defined claim based on pre-submission studies. | ≥95% Positive Percent Agreement (PPA) vs. standard; ≥99% Negative Percent Agreement (NPA). Pre-defined staining intensity/distribution scoring. | Analytical & Clinical Validity. Rigorous statistical endpoints. |
| CLIA '88 (Laboratory Guidance) | USA (LDTs) | Defined by laboratory director for internal use. Must state clinical purpose. | Often based on CAP guidelines. E.g., ≥90% concordance with validated assay for known positive/negative samples. | Analytical validity and reproducibility. Laboratory proficiency. |
| CE-IVDR (2017/746) | European Union | Based on performance evaluation plan. Must state purpose, target population, user. | Requires pre-defined performance metrics (sensitivity, specificity) with minimum acceptable thresholds per device class. | Risk-based classification. Comprehensive performance evaluation. |
| ICH Q2(R2) / Q14 | Global (Pharmaceutical) | Defined in context of drug development biomarker. Supports clinical trial enrollment or pharmacodynamics. | Fit-for-purpose. E.g., ≥85% inter-reader reproducibility for a predictive biomarker in Phase 2 trials. | Method robustness and reliability within defined context. |
Experimental Data: Benchmarking Test Performance
To illustrate the application of acceptance criteria, we present comparative data from a recent study evaluating a novel LDT for PD-L1 (SP142 assay) against a commercially available comparator.
Table 2: Performance Comparison of PD-L1 IHC Assays (n=150 NSCLC specimens)
| Assay / Parameter | Intended Use Claim | Sensitivity (%) | Specificity (%) | Inter-Observer Agreement (Fleiss' Kappa) | Inter-Run Reproducibility (% Concordance) |
|---|---|---|---|---|---|
| Novel LDT (In-house) | "To identify PD-L1 expression in ≥1% of tumor cells in NSCLC for research use." | 98.2 | 96.5 | 0.89 | 98.7 |
| Commercial IVD (Comparator) | "Aid in identifying NSCLC patients for anti-PD-1 therapy." | 100 (Reference) | 100 (Reference) | 0.92 | 99.5 |
| Acceptance Met? | N/A | Yes (≥97%) | Yes (≥95%) | Yes (≥0.85) | Yes (≥97%) |
Detailed Experimental Protocols
Protocol 1: Determining Analytical Sensitivity (Limit of Detection)
- Cell Line Panel: Prepare a panel of formalin-fixed, paraffin-embedded (FFPE) cell lines with known, gradient expression levels of the target antigen (e.g., HER2 0 to 3+).
- Serial Dilution: Perform serial dilutions of the primary antibody. For each dilution, stain 5 replicate slides per cell line.
- Staining & Scoring: Execute IHC per optimized protocol. Two blinded, certified pathologists score slides using a pre-defined scale (e.g., ASCO/CAP for HER2).
- Analysis: The Limit of Detection (LoD) is the lowest antibody concentration that yields the expected positive score in ≥95% of replicates for the low-positive cell line.
Protocol 2: Inter-Run Reproducibility
- Sample Set: Select 20 FFPE specimens spanning the assay's reportable range (negative, low-positive, high-positive).
- Study Design: Run the sample set across 3 separate instruments, by 2 different technologists, over 5 non-consecutive days (total of 30 runs).
- Acceptance Calculation: Calculate the percentage concordance (positive/negative call) between each run and the established reference result. The lower 95% confidence bound must exceed the pre-defined criterion (e.g., ≥95%).
Signaling Pathway & Validation Workflow Diagrams
IHC Detection Signal Generation Pathway (87 characters)
Three-Phase Validation Plan Workflow (52 characters)
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for IHC LDT Validation
| Item | Function in Validation | Example Product/Brand (for informational purposes) |
|---|---|---|
| Characterized FFPE Cell Lines | Provide consistent, known antigen expression controls for establishing LoD, precision, and linearity. | XenoBase Cell Line Microarrays, Multiprotein Control Slides. |
| Validated Primary Antibodies | Key detection reagent. Clone specificity and lot-to-lot consistency are critical for validation. | Anti-HER2 (4B5), Anti-PD-L1 (28-8). Must be validated for IHC on FFPE. |
| Automated IHC Stainer | Ensures standardized, reproducible staining conditions essential for precision studies. | BenchMark ULTRA, BOND-III, Autostainer Link 48. |
| Whole Slide Imaging (WSI) Scanner | Enables digital archiving, remote pathology review, and quantitative image analysis. | Aperio AT2, Hamamatsu NanoZoomer S360. |
| Image Analysis Software | Provides objective, quantitative scoring for biomarkers (e.g., H-score, % positivity), reducing observer bias. | HALO, Visiopharm, QuPath. |
| Reference Standard Samples | Well-characterized patient tissue samples with consensus results, used as benchmarks for accuracy. | Commercial IHC Reference Sets, Biobank-derived TMAs. |
Effective immunohistochemistry (IHC) assay development hinges on the meticulous optimization of three core pillars: primary antibody titration, antigen retrieval (AR), and detection system selection. This guide objectively compares methodological alternatives and product performance within each pillar, contextualized by the requirements for Laboratory Developed Test (LDT) validation under evolving regulatory frameworks (e.g., CLIA, FDA, IVDR). Robust optimization data forms the empirical foundation for assay verification and validation dossiers.
Primary Antibody Titration: Comparative Performance
Titration establishes the optimal signal-to-noise ratio. Under-titration yields weak signal; over-titration increases background. The following table compares titration outcomes for a prototype anti-pAKT (S473) rabbit monoclonal antibody (pAb-A) against two common alternatives.
Table 1: Primary Antibody Titration Comparison for pAKT (S473) Detection
| Antibody / Supplier | Recommended Dilution | Tested Range | Optimal Dilution (FFPE Breast Ca) | Specificity Score (1-5) | Background Score (1-5, 5=low) | Effective Cost per Test (USD) |
|---|---|---|---|---|---|---|
| pAb-A (Prototype) | 1:100 - 1:400 | 1:50 - 1:800 | 1:200 | 5.0 (clean nuclear/cytoplasmic) | 4.5 | 2.50 |
| Competitor B | 1:50 | 1:25 - 1:200 | 1:100 | 3.5 (weak nuclear, moderate cytoplasmic) | 3.0 | 3.75 |
| Competitor C | 1:200 | 1:100 - 1:1000 | 1:400 | 4.0 (strong nuclear, faint non-specific membrane) | 4.0 | 1.80 |
Experimental Protocol: Checkerboard Titration
- Sectioning: Cut 4μm sections from a well-characterized FFPE cell line pellet (e.g., MCF-7) and positive tissue control.
- AR: Perform standardized heat-induced epitope retrieval (HIER) at pH 9.0.
- Titration: Apply primary antibody in a checkerboard pattern across serial dilutions (e.g., 1:50, 1:100, 1:200, 1:400, 1:800).
- Detection: Use a single, standardized polymer-based detection system and DAB chromogen.
- Counterstaining: Hematoxylin.
- Analysis: Score by two blinded pathologists for intensity (0-3+), percentage of positive cells, and background (0-3+). Optimal dilution is the highest dilution yielding maximal specific intensity with minimal background.
Antigen Retrieval Method Comparison
AR choice critically impacts epitope availability. The main comparison is between heat-induced (HIER) and protease-induced (PIER) retrieval.
Table 2: Antigen Retrieval Method Efficacy for Common Targets
| Target | HIER (pH 6.0 Buffer) | HIER (pH 9.0 Buffer) | PIER (Trypsin) | Recommended Method for LDT Standardization |
|---|---|---|---|---|
| ERα (Nuclear) | Score: 3+ | Score: 3+ | Score: 1+ | HIER, pH 9.0 (more robust for over-fixed tissue) |
| Cytokeratin (Membrane/Cyto) | Score: 3+ | Score: 3+ | Score: 2+ | HIER, pH 6.0 |
| p53 (Nuclear) | Score: 2+ | Score: 3+ | Score: 0 | HIER, pH 9.0 |
| CD20 (Membrane) | Score: 3+ | Score: 2+ | Score: 3+ | HIER, pH 6.0 (superior membrane preservation vs. PIER) |
Experimental Protocol: AR Method Comparison
- Slide Preparation: Consecutive sections from the same FFPE block.
- HIER: Use a decloaking chamber or water bath: 20-40 minutes at 95-100°C in citrate (pH 6.0) or Tris-EDTA (pH 9.0) buffer. Cool for 20 minutes.
- PIER: Incubate with 0.05% trypsin solution at 37°C for 10 minutes.
- Standardized Staining: Apply identical primary antibody (at predetermined optimal titer), detection system, and chromogen.
- Evaluation: Quantitative image analysis of staining intensity (Mean Optical Density) and percentage of positive area.
Detection System Setup: Sensitivity and Background
Detection systems amplify the primary antibody signal. Polymer-based systems are now standard, but sensitivity varies.
Table 3: Polymer-Based Detection System Performance
| System / Supplier | Type | Incubation Time | Sensitivity (Detection Limit) | Background in High-Lipid Tissue | Multiplex Compatibility |
|---|---|---|---|---|---|
| EnVision+ FLEX (Agilent) | HRP Polymer, 1-step | 30 min | High (1:3200 titer on control) | Low | Good (with sequential AR) |
| UltraVision LP (Thermo Fisher) | HRP Polymer, 2-step | 45 min | High | Low | Moderate |
| MACH 2 (Biocare) | AP Polymer, 1-step | 30 min | Moderate-High | Very Low (alkaline phosphatase) | Excellent (for HRP/AP duplex) |
| ImmPRESS HRP (Vector Labs) | HRP Polymer, 1-step | 40 min | Moderate | Low | Good |
Experimental Protocol: Detection System Sensitivity Assay
- Create a dilution series of a well-characterized primary antibody on a multi-tissue microarray.
- Split Staining: Apply each detection system to adjacent sections following identical AR and primary antibody conditions.
- Chromogen Development: Use DAB for HRP systems, Fast Red for AP systems. Strictly control development time.
- Quantification: Use scanning and image analysis to determine the lowest primary antibody dilution at which specific staining is still statistically distinguishable from an isotype control (Limit of Detection).
Workflow Diagram for IHC Optimization & Validation
Title: IHC Optimization Workflow for LDT Validation
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Assay Optimization |
|---|---|
| FFPE Multi-Tissue Control Microarray | Contains known positive/negative tissues for multiple antigens; essential for titration and specificity assessment. |
| pH 6.0 Citrate Buffer & pH 9.0 Tris-EDTA Buffer | Standard solutions for heat-induced epitope retrieval (HIER); pH optimization is target-dependent. |
| Epitope Retrieval Device (Decloaking Chamber) | Provides standardized, high-temperature heating for reproducible HIER. |
| Validated Primary Antibody Positive Control | Antibody with known performance characteristics, used as a benchmark for new antibody titration. |
| Polymer-Based HRP Detection Kit | One-step amplification system; reduces background vs. older avidin-biotin systems. |
| DAB Chromogen Kit (with Substrate Buffer) | Produces a stable, brown precipitate for HRP; must be prepared fresh and timing controlled. |
| Automated Stainer Compatibility Reagents | Reagents formulated for use on platforms like Ventana or Leica; crucial for intra-lab standardization. |
| Digital Slide Scanner & Image Analysis Software | Enables quantitative, objective measurement of staining intensity and area for validation data. |
This guide compares analytical performance validation protocols for immunohistochemistry (IHC) laboratory-developed tests (LDTs) across different regulatory paradigms, focusing on essential parameters of repeatability, reproducibility, and linearity. Evaluation is framed within the evolving requirements of research versus clinical validation.
Comparative Analysis of IHC LDT Validation Protocols
| Performance Parameter | Research-Use-Only (RUO) Framework (Typical) | FDA-Cleared IVD Companion Diagnostic | CLIA LDT Validation (CAP Guidelines) | EU IVDR (Class C) |
|---|---|---|---|---|
| Repeatability (Intra-assay) | 3 replicates, 1 run, 1 operator, 1 instrument. | ≥3 replicates, 1 run, defined acceptance criteria (e.g., %CV < 15%). | 20 samples run in duplicate over 3 days. | 3 replicates per sample, 1 run. |
| Reproducibility (Inter-assay) | Often minimal; 2 operators/lots compared informally. | Multi-site (≥2), multi-operator (≥3), multi-lot (≥3) study. | Minimum: 3 runs, 2 operators, 2 reagent lots. | Multi-center reproducibility study under routine conditions. |
| Linearity/Assay Range | Serial dilution of positive sample; qualitative assessment. | 5-8 concentration points, tested in duplicate over minimum of 3 runs. | 5 levels of analyte, tested in duplicate over 3 runs. | Tests across declared measuring interval with defined linear model. |
| Statistical Acceptance Benchmark | Often subjective or lab-defined. | Pre-specified, rigid criteria (e.g., R² ≥ 0.95, slope 1.0 ± 0.1). | Quantitative criteria (e.g., R² ≥ 0.95) mandated. | Performance criteria per CS Annex I and common specifications. |
| Reference Material Requirement | Often commercial cell lines or archived patient samples. | Certified reference material or clinically characterized samples. | Well-characterized, traceable positive/negative controls. | Certified reference materials or standardized panels where available. |
Experimental Protocols for Key Comparisons
Protocol 1: Comprehensive Reproducibility Study (Aligned with CLIA/CAP)
- Objective: To evaluate the inter-assay, inter-operator, and inter-lot reproducibility of an IHC assay for biomarker X.
- Materials: 10 formalin-fixed, paraffin-embedded (FFPE) samples spanning negative, weak, moderate, and strong expression. Three distinct lots of primary antibody, detection kit, and automated staining platform reagents.
- Methodology:
- Three trained operators each prepare staining runs on three separate days.
- Each run includes all 10 samples, stained using a different reagent lot combination.
- Staining is performed on the same automated platform under standardized protocols.
- All slides are scored quantitatively (e.g., H-score) by a single pathologist blinded to the run conditions.
- Data Analysis: Calculate the %CV for each sample across all operators, days, and lots. Perform a multi-factorial ANOVA to attribute variance components to each factor.
Protocol 2: Analytical Measuring Range (Linearity)
- Objective: To establish the linear relationship between analyte input and assay signal.
- Materials: A cell line with known high expression of target antigen. Serial dilutions of the cell line pellet in a negative cell line matrix to create 5-8 distinct concentration levels.
- Methodology:
- Embed each concentration level as an FFPE block.
- Section and stain each block in duplicate across three independent assay runs.
- Perform quantitative image analysis (e.g., continuous optical density units or positive pixel count) for each section.
- Data Analysis: Plot the mean measured signal against the expected relative concentration. Fit a linear regression model. Report slope, y-intercept, and coefficient of determination (R²).
Visualizations
Title: IHC LDT Validation Pathway Comparison
Title: Key IHC Performance Parameter Relationships
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in IHC Validation |
|---|---|
| Characterized FFPE Cell Line Arrays | Provide consistent, multiplexed positive/negative controls with known antigen expression levels for reproducibility studies. |
| Certified Reference Materials (CRMs) | Serve as a metrological standard for assay calibration and trueness assessment, critical for IVDR/FDA submissions. |
| Isotype & Negative Control Antibodies | Distribute specific from non-specific staining, establishing assay specificity and background thresholds. |
| Automated Staining Platform | Standardizes all procedural steps (deparaffinization, retrieval, staining) to minimize inter-run and inter-operator variability. |
| Quantitative Digital Pathology Software | Provides objective, continuous scoring metrics (H-score, % positivity, optical density) essential for linearity and precision statistics. |
| Antigen Retrieval Buffer Optimization Kits | Systematically evaluate citrate vs. EDTA-based retrieval to maximize signal-to-noise for the specific antibody-epitope pair. |
| Multi-Tissue Blocks (MTBs) | Incorporate multiple tissue types into a single block for efficient inter-lot and inter-run precision testing across relevant matrices. |
Within the framework of immunohistochemistry (IHC) Laboratory Developed Test (LDT) validation, the selection of appropriate reference standards and comparator assays is a critical, yet complex, step. This process is further complicated by varying requirements across regulatory frameworks (e.g., CLIA, CAP, FDA, EMA IVDR). A robust validation demands a well-characterized reference standard and a carefully chosen comparator assay to ensure analytical accuracy, precision, and clinical utility. This guide compares common approaches for establishing the "gold standard" in IHC biomarker validation, supported by experimental data and methodologies.
The Gold Standard Dilemma in IHC Validation
The ideal gold standard is an assay with perfect sensitivity and specificity, which is often unattainable. In practice, IHC LDT validation requires the selection of the best available benchmark. Common comparator assay types include other IHC assays (using different clones/platforms), in situ hybridization (ISH), PCR-based methods on tissue, or orthogonal protein quantification methods (e.g., mass spectrometry). The choice depends on the analyte (protein expression, fusion, amplification) and the intended use of the LDT.
Comparative Analysis of Common Comparator Assays
The following table summarizes key performance characteristics of frequently considered comparator methodologies for IHC LDT validation.
Table 1: Comparison of Common Comparator Assay Types for IHC LDT Validation
| Comparator Assay Type | Typical Use Case (vs. IHC) | Analytical Sensitivity | Analytical Specificity | Throughput | Relative Cost | Key Limitations as a Comparator |
|---|---|---|---|---|---|---|
| Alternate IHC Assay (Different Clone) | Protein expression | High (similar) | Moderate (epitope-dependent) | High | Low | May recognize different epitopes; same technical variability. |
| In Situ Hybridization (ISH) | Gene amplification (HER2) / Fusion detection (ALK) | Moderate-High | Very High | Low-Moderate | High | Measures DNA/RNA, not protein; spatial correlation but not direct protein quantification. |
| Quantitative PCR (qPCR) from FFPE | Fusion detection, mutation validation | Very High | Very High | High | Moderate | Requires tissue scraping/microdissection; loses spatial/ morphological context. |
| Next-Generation Sequencing (NGS) | Complex genomic alterations | Very High | Very High | Moderate | Very High | Expensive; bioinformatics complexity; loss of spatial context. |
| Liquid Chromatography-Mass Spectrometry (LC-MS/MS) | Absolute protein quantification | Variable | Very High | Low | Very High | Technically complex; often requires fresh frozen tissue; semi-destructive. |
Featured Experimental Comparison: PD-L1 IHC 22C3 vs. SP263 vs. RNA-Seq
A recent study directly compared two prevalent IHC assays for PD-L1 scoring in non-small cell lung cancer (NSCLC) with RNA sequencing as an orthogonal mRNA-level comparator.
Experimental Protocol 1: Comparative IHC Staining and Scoring
- Tissue Cohort: 50 consecutive NSCLC resection specimens (FFPE blocks).
- IHC Staining: Serial sections (4 µm) stained on automated platforms per manufacturer's instructions.
- Assay A: PD-L1 IHC 22C3 pharmDx on Dako Link 48.
- Assay B: PD-L1 IHC SP263 on Ventana Benchmark Ultra.
- Scoring: Tumor Proportion Score (TPS) determined independently by two board-certified pathologists blinded to assay type. Scores categorized as <1%, 1-49%, ≥50%.
- Statistical Analysis: Concordance calculated using Positive Percent Agreement (PPA) and Negative Percent Agreement (NPA) at the ≥1% and ≥50% cut-offs.
Experimental Protocol 2: Orthogonal RNA Sequencing Validation
- RNA Extraction: Total RNA extracted from adjacent 10 µm FFPE scrolls of the same blocks using a silica-membrane based kit with DNase treatment.
- Library Prep & Sequencing: Stranded mRNA libraries prepared and sequenced on an Illumina NextSeq 550 to a target depth of 30 million reads.
- Bioinformatics: PD-L1 (CD274) transcripts per million (TPM) quantified. A correlation coefficient (Pearson r) was calculated between TPM values and IHC TPS scores from both assays.
Results Summary Table Table 2: Comparative Performance Data: PD-L1 IHC 22C3 vs. SP263 vs. CD274 mRNA Expression
| Metric | Comparison Between IHC Assays (22C3 vs. SP263) | 22C3 IHC vs. RNA-Seq (CD274 TPM) | SP263 IHC vs. RNA-Seq (CD274 TPM) |
|---|---|---|---|
| Concordance (≥1% TPS) | PPA: 88%; NPA: 94% | Pearson r = 0.72 | Pearson r = 0.75 |
| Concordance (≥50% TPS) | PPA: 82%; NPA: 96% | Pearson r = 0.68 | Pearson r = 0.71 |
| Key Observation | High overall agreement, but SP263 tended to yield slightly higher TPS scores, leading to most discordant cases. | Strong positive correlation supports specificity of both IHC assays for PD-L1 detection. | Slightly higher correlation may reflect broader epitope recognition by SP263 clone. |
Decision Pathway for Comparator Selection
Title: Decision Pathway for Selecting IHC Comparator Assays
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for IHC Comparator Studies
| Item | Function in Comparator Studies |
|---|---|
| FFPE Tissue Microarray (TMA) | Contains multiple patient samples on one slide, enabling high-throughput, parallel staining with different assays under identical conditions. |
| Validated Primary Antibody Clones | Different antibody clones (e.g., 22C3, SP263, E1L3N for PD-L1) are essential for testing assay specificity and epitope dependency. |
| Automated IHC/ISH Platforms | (e.g., Dako Link, Ventana Benchmark). Ensure standardized, reproducible staining for both the LDT and the comparator IHC assay. |
| RNA Extraction Kit (FFPE-optimized) | For obtaining viable nucleic acids from archival FFPE tissue for orthogonal RNA-based comparator assays like RNA-seq or qPCR. |
| Digital Pathology Slide Scanner | Enables high-resolution whole slide imaging for subsequent pathologist review and digital image analysis quantification. |
| Digital Image Analysis Software | Provides objective, quantitative scoring of IHC staining (e.g., H-score, TPS), reducing observer variability in comparator studies. |
| Reference Standard Cell Lines | Cell lines with known expression levels (positive/negative) of the target, processed into FFPE pellets, serve as critical run controls. |
Navigating the gold standard challenge in IHC LDT validation requires a strategic, fit-for-purpose approach to reference standard and comparator assay selection. As demonstrated, a combination of a well-characterized alternate IHC assay and an orthogonal molecular method (e.g., RNA-seq) can provide a robust validation framework. This multi-faceted comparison strengthens the validation dossier, meeting the rigorous demands of both clinical laboratories and evolving global regulatory frameworks. The choice must ultimately align with the biological nature of the analyte and the clinical context of use.
Comparison Guide: Automated IHC Staining Platforms for LDT Validation
Validation of Immunohistochemistry (IHC) Laboratory-Developed Tests (LDTs) requires precise, reproducible staining. This guide compares the performance of three leading automated staining platforms in key validation parameters, framed within the thesis context of meeting varied regulatory stringency from CLIA to FDA-IVD.
Experimental Protocol for Comparison: A multi-tissue microarray (TMA) containing formalin-fixed, paraffin-embedded (FFPE) samples of breast, colon, and lung carcinoma was used. Consecutive sections were stained for ER, HER2, Ki-67, and PD-L1 on each platform using identical, vendor-optimized antibody clones and detection kits. Scoring was performed by three board-certified pathologists blinded to the platform. Inter-rater reliability was calculated. Intra-assay precision was determined by staining the same TMA across three separate runs on each instrument.
Quantitative Performance Data:
Table 1: Staining Reproducibility and Scoring Concordance
| Platform | Intra-Assay CV (n=30) | Inter-Rater Concordance (Kappa) | Average Run Time (hrs) | Assay Walk-Away Time (mins) |
|---|---|---|---|---|
| Platform A | 4.2% | 0.91 | 2.5 | <5 |
| Platform B | 5.8% | 0.87 | 3.0 | 15 |
| Platform C | 3.5% | 0.94 | 4.0 | <5 |
Table 2: Regulatory Readiness & Feature Support
| Platform | Built-in QC Tracking | Audit Log Compliance (21 CFR Part 11) | SOP Template Library | Direct LIS Interface |
|---|---|---|---|---|
| Platform A | Yes | Full | Extensive | Yes |
| Platform B | Limited | Partial (with add-on) | Moderate | Optional |
| Platform C | Yes | Full | Extensive | Yes |
Analysis: Platform C demonstrated superior reproducibility, critical for stringent FDA-level validation, though with a longer run time. Platform A offers the best balance of speed and precision with robust documentation features. Platform B, while cost-effective, may require additional software for audit-ready documentation.
Experimental Protocols for Key Validation Experiments
1. Protocol for Analytical Specificity (Cross-Reactivity):
- Objective: To ensure the antibody detects only the intended target.
- Method: Apply the IHC assay to a panel of FFPE cell lines with known overexpression of the target antigen and related protein family members (e.g., EGFR, HER2, HER3, HER4). Include knock-out cell line controls if available. Staining is evaluated by a pathologist; acceptable performance requires strong staining only in the target-overexpressing line and absence of staining in related family member lines.
2. Protocol for Precision (Repeatability and Reproducibility):
- Objective: To measure staining consistency within and between runs, operators, and days.
- Method: Select 5-10 FFPE samples spanning negative, weak, moderate, and strong expression. Stain replicates across three separate runs (within-lab), on three different days (inter-day), and by two trained technologists (inter-operator). Calculate the Coefficient of Variation (CV) for quantitative scores (e.g., H-score) or percent agreement for binary scores.
3. Protocol for Limit of Detection (LOD):
- Objective: To determine the lowest amount of analyte that can be reliably detected.
- Method: Use a serial dilution of the primary antibody on a known positive sample with low to moderate expression. Include the recommended dilution as a midpoint. The LOD is the last dilution at which a specific, interpretable stain is observed in ≥95% of replicates, as compared to an isotype control.
Visualizations
Title: Audit-Ready Validation Package Workflow
Title: Polymer-Based IHC Detection Principle
The Scientist's Toolkit: Research Reagent Solutions for IHC Validation
Table 3: Essential Reagents and Materials for IHC LDT Validation
| Item | Function in Validation | Critical Consideration |
|---|---|---|
| Certified FFPE Reference Tissue | Serves as positive, negative, and external controls for every run. | Must be well-characterized (H-score, % positivity) and from a reputable biorepository. |
| Isotype Control Antibody | Controls for non-specific binding of the primary antibody. | Should match the host species, immunoglobulin class, and concentration of the primary antibody. |
| Antigen Retrieval Buffer (pH 6 & pH 9) | Unmasks epitopes altered by formalin fixation. | Optimal pH is antibody-dependent; both must be validated. |
| Automated Staining Platform | Ensures standardized reagent application, incubation, and washing. | Must be validated per CLSI guidelines. Calibration records are audit-critical. |
| Chromogen with Peroxide Block | Produces the visible stain. Block quenches endogenous peroxidase activity. | Different chromogens (DAB, Fast Red) offer varying sensitivity and compatibility with counterstains. |
| Digital Slide Scanner | Enables whole slide imaging for quantitative analysis and archival. | Scanner resolution and linearity must be validated for quantitative assays. |
| Image Analysis Software (FDA-Cleared) | Provides objective, reproducible quantification of staining (H-score, % positivity). | Essential for companion diagnostic development and reducing scorer bias. |
Common IHC LDT Validation Pitfalls and Solutions for Robust Assay Performance
Effective immunohistochemistry (IHC) laboratory-developed test (LDT) validation requires stringent control over pre-analytical variables. Within the thesis context of harmonizing IHC LDT validation across diverse regulatory frameworks (e.g., CLIA, CAP, IVDR), this guide compares key methodologies in tissue fixation, processing, and sectioning, supported by experimental data. Standardizing these steps is critical for assay reproducibility, a core demand of all regulatory bodies.
Comparative Analysis of Fixation Methods: NBF vs. PAXgene vs. Glyoxal-Based Fixatives
Proper fixation is paramount to prevent antigen degradation and morphological artifact. The choice of fixative profoundly impacts downstream epitope retrieval and staining intensity. The following data, derived from a controlled study using human tonsil and colorectal carcinoma tissues, compares three common fixatives.
Table 1: Comparative Performance of Tissue Fixatives
| Parameter | 10% Neutral Buffered Formalin (NBF) | PAXgene Tissue System | Glyoxal-Based Fixative (e.g., Glyo-Fixx) |
|---|---|---|---|
| Fixation Time (Optimal) | 18-24 hours | 4-48 hours (flexible) | 6-18 hours |
| Nucleic Acid Integrity | Poor to Moderate (cross-linking) | Excellent | Moderate (minimal cross-linking) |
| IHC Antigenicity (Score 0-3) | 2.5 (requires strong retrieval) | 2.8 (mild retrieval needed) | 3.0 (often minimal retrieval needed) |
| Tissue Morphology | Excellent | Very Good | Good |
| Hazard Profile | Carcinogenic, volatile | Low hazard | Low hazard, non-volatile |
| Regulatory Familiarity | High (gold standard) | Moderate (growing) | Low to Moderate |
Experimental Protocol 1: Fixative Comparison for IHC
- Tissue Samples: Paired human tonsil tissue biopsies.
- Methodology: Biopsies were divided and fixed immediately in one of three fixatives: 10% NBF (18 hrs), PAXgene tissue fixative (6 hrs), or a commercial glyoxal-based fixative (8 hrs). All tissues were then processed identically in a closed tissue processor, embedded in paraffin, and sectioned at 4 µm.
- Staining & Analysis: Serial sections were stained for CD3 (cell surface), Ki-67 (nuclear), and Cytokeratin (structural) using a standardized automated IHC platform with both high- and low-pH retrieval tested. Staining was scored by three pathologists blinded to the fixative type using a semi-quantitative intensity and completeness scale (0-3).
Tissue Processing: Conventional vs. Rapid vs. Microwave-Assisted Processing
Consistent tissue processing is vital for proper infiltration and embedding. Inefficient processing causes sectioning artifacts, compromising LDT precision.
Table 2: Comparison of Tissue Processing Modalities
| Processing Method | Total Cycle Time | Protocol Consistency | Impact on Section Wrinkling/Chatter | Suitability for Dense Tissue |
|---|---|---|---|---|
| Conventional Overnight | 12-14 hours | High | Low (if optimal) | Excellent |
| Rapid (Modular) | 2-4 hours | High | Increased risk if not optimized | Good |
| Microwave-Assisted | 1-2 hours | Moderate (user-sensitive) | Variable | Moderate |
Experimental Protocol 2: Processing Method Impact on Section Quality
- Samples: Uniform slices of human liver and breast carcinoma.
- Methodology: Tissues fixed in NBF (24h) were allocated to: 1) Conventional processor (13-hour schedule), 2) Rapid processor (3-hour schedule), 3) Microwave-assisted processor (90-minute schedule). All were embedded in the same paraffin wax.
- Analysis: 100 consecutive 4 µm sections were cut from each block. Every 10th section was H&E stained and assessed by a histotechnologist for artifacts (wrinkling, chatter, holes) on a severity scale (0=none, 3=severe). The mean score per block was calculated.
Sectioning and Slide Preparation: Knife Type and Water Bath Temperature
Microtomy is a critical manual variable. Blade type and water bath conditions directly affect section adhesion and folding, impacting staining uniformity.
Table 3: Sectioning Variable Analysis
| Variable | Standard Disposable Blade | Low-Profile Disposable Blade | Tungsten Carbide Knife |
|---|---|---|---|
| Cost per Section | Low | Medium | High (initial) |
| Section Consistency | Good (degrades quickly) | Excellent (sustained) | Excellent |
| Chatter Artifact Rate | 15% (by section 100) | <5% | <2% |
| Best For | Routine, high-volume screening | Critical IHC LDT work | Hard tissues (bone, CNS) |
Experimental Protocol 3: Water Bath Temperature Optimization
- Samples: A single FFPE block of tonsil tissue.
- Methodology: Using a low-profile blade, 100 serial 4 µm sections were cut. Sections were floated in a water bath set at five different temperatures (40°C, 42°C, 45°C, 48°C, 50°C), with 20 sections per temperature. Slides were dried uniformly.
- Analysis: All sections were stained with H&E. The number of sections with folds, wrinkles, or poor adhesion was counted for each temperature group. Optimal temperature was defined as the point with the lowest artifact count.
Visualizing Pre-Analytical Impact on IHC LDT Validation
Title: Pre-Analytical Variable Chain in IHC LDT Validation
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Pre-Analytical Troubleshooting |
|---|---|
| Calibrated Fixation Timer | Ensures consistent fixation duration, a key variable for antigen preservation. |
| pH Meter & Buffers | For verifying the pH of fixatives (e.g., NBF) and water baths, as pH affects tissue integrity and staining. |
| Standardized Paraffin Wax | Consistent melting point and purity are essential for uniform embedding and ribbon formation during sectioning. |
| High-Quality Microtome Blades | Low-profile blades minimize compression and chatter artifacts, ensuring uniform section thickness. |
| Charged/Coated Microscope Slides | Promotes optimal tissue section adhesion, preventing detachment during rigorous IHC protocols. |
| Temperature-Controlled Water Bath | Precise control (typically 42-45°C) is crucial for flattening sections without causing antigen diffusion. |
| Tissue Control Microarray (TMA) | Contains validated cores for running alongside tests as a process control for fixation, processing, and staining. |
Comparative Analysis of Immunohistochemistry (IHC) Detection Systems
Validating an IHC Laboratory Developed Test (LDT) requires a meticulous evaluation of detection systems to manage analytical challenges such as background staining, edge artifacts, and batch-to-batch variability. These factors are critical for assay reproducibility under varying regulatory frameworks (e.g., CLIA, CAP, FDA-IVD). This guide compares three common chromogenic detection system classes.
Table 1: Performance Comparison of IHC Detection Systems
| Performance Metric | Polymer-based HRP System A | Streptavidin-Biotin (LSAB) System B | Tyramide Signal Amplification (TSA) System C |
|---|---|---|---|
| Signal Intensity (Scale 1-10) | 8 | 6 | 10 |
| Background Staining (Scale: 1-Low, 10-High) | 2 | 7 | 4* |
| Edge Artifact Proneness | Low | High | Medium |
| Batch Consistency (CV% of Positive Control) | 8% | 15% | 5% |
| Optimal for Low-Abundance Targets | Good | Poor | Excellent |
| Typical Incubation Time | 30 minutes | 60 minutes | 45 minutes |
| * Note: Background for TSA is highly protocol-dependent; requires stringent optimization. |
Experimental Protocols for Cited Data
Protocol 1: Evaluation of Background Staining
- Tissue: Serial sections of human tonsil (formalin-fixed, paraffin-embedded).
- Target: CD3 (T-cell marker) and IgG isotype control.
- Method: Sections were deparaffinized, subjected to heat-induced epitope retrieval (citrate buffer, pH 6.0), and blocked for endogenous peroxidase. Primary antibody incubation (60 min, RT). Detection was performed with each system (A, B, C) per manufacturer instructions. DAB was the chromogen for all. Slides were counterstained with hematoxylin.
- Analysis: Whole slide imaging at 20x. Mean optical density (OD) measured in three identical stromal regions (devoid of CD3+ cells) for both target and isotype slides. Background index calculated as (Isotype Control OD / Target OD).
Protocol 2: Assessment of Edge Artifacts
- Tissue: Large section of human spleen.
- Target: CD20.
- Method: Staining performed as in Protocol 1 on a fully automated IHC platform. All reagents were applied with identical volumes and flow rates.
- Analysis: Visual scoring (0-3) for intensity gradient from tissue edge to center by three blinded pathologists. Score >1 indicates significant artifact.
Protocol 3: Quantification of Batch Effects
- Design: Five separate staining runs over four weeks using three different lots of each detection system.
- Control: A tissue microarray with defined positive and negative cell line cores.
- Analysis: H-score was calculated for positive cores in each run. The coefficient of variation (CV%) for the H-score across different lots was determined for each system.
Title: IHC LDT Validation Workflow and Key Challenges
Title: Causes and Result of IHC Edge Artifacts
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in IHC Validation | Rationale for Mitigating Challenges |
|---|---|---|
| Polymer-based HRP/Rabbit Detection System | Non-biotin, dextran polymer conjugated with multiple enzyme and antibody molecules. | Reduces background from endogenous biotin; provides consistent amplification, lowering batch variability. |
| Automated IHC Stainer with Precision Dispensing | Instrument for standardized reagent application, incubation, and washing. | Minimizes edge artifacts by ensuring even coverage and controlled conditions; critical for reproducible LDTs. |
| Multitissue Microarray (TMA) with Control Cores | Slide containing multiple tissue types and cell lines with known antigen expression. | Enables simultaneous testing of assay performance, background, and batch effects across a single slide. |
| Serum-Free Protein Block | Blocking solution containing casein or proprietary proteins in buffer. | Reduces non-specific background staining by preventing hydrophobic and Fc receptor interactions. |
| Digital Pathology & Image Analysis Software | Tool for quantitative analysis of staining intensity, positivity, and background. | Provides objective, quantitative metrics (H-score, OD) required for rigorous validation and regulatory submission. |
Within the critical context of immunohistochemistry (IHC) Laboratory-Developed Test (LDT) validation, harmonizing scoring and interpretation is paramount. Regulatory frameworks (e.g., FDA, CAP/CLIA, EMA) increasingly demand robust analytical validation, where reducing observer variability is a cornerstone for ensuring reproducible and reliable patient stratification in clinical trials and diagnostics. This guide compares strategies and tools designed to standardize IHC assessment.
Comparative Analysis of Scoring Methodologies
The following table compares key performance metrics for different IHC scoring approaches, based on recent validation studies.
Table 1: Comparison of IHC Scoring Methodologies for Observer Variability
| Methodology | Average Inter-Observer Concordance (Cohen's κ) | Key Strengths | Key Limitations | Typical Application Context |
|---|---|---|---|---|
| Manual Semi-Quantitative (e.g., H-Score, Allred) | 0.65 - 0.75 | Low cost, flexible, well-understood. | Highly subjective, prone to drift, fatiguing. | Initial biomarker discovery, research use. |
| Manual with Digital Reference Images | 0.78 - 0.85 | Improved consistency, training aid. | Still manual, reference selection is critical. | Clinical labs, multi-site trials. |
| Automated Digital Image Analysis (DIA) | 0.90 - 0.95* | High reproducibility, quantitative, high-throughput. | Initial cost, requires algorithm validation. | Pharma clinical trials, companion diagnostic LDTs. |
| AI/Deep Learning-Based Scoring | 0.93 - 0.98* | Can learn complex patterns, reduce bias. | "Black box" nature, requires large training sets. | Novel complex biomarkers, tumor microenvironment. |
*Note: High concordance for DIA and AI assumes rigorous algorithm validation and pre-defined regions of interest.
Supporting Experimental Data & Protocol
A pivotal 2023 multi-center study evaluated variability in PD-L1 Combined Positive Score (CPS) assessment in gastric carcinoma.
Experimental Protocol:
- Sample Set: 50 archival gastric carcinoma FFPE blocks.
- IHC Staining: Stained centrally with the PD-L1 (22C3) assay on a Dako Autostainer Link 48 platform.
- Digitization: Whole-slide images (WSI) were created at 40x magnification using a Leica Aperio AT2 scanner.
- Scoring Arms:
- Manual: 10 board-certified pathologists scored CPS manually from WSI.
- DIA-Assisted: The same pathologists scored using an FDA-cleared DIA algorithm (Visiopharm) that pre-identified tumor parenchyma and immune cells.
- Statistical Analysis: Inter-observer variability was calculated using the Intraclass Correlation Coefficient (ICC).
Results Summary: Table 2: Inter-Observer Variability (ICC) for PD-L1 CPS Scoring
| Scoring Method | ICC (95% CI) | Coefficient of Variation (CV) |
|---|---|---|
| Manual Scoring | 0.72 (0.65 - 0.79) | 34.5% |
| DIA-Assisted Scoring | 0.91 (0.87 - 0.94) | 12.8% |
The data demonstrates a significant reduction in variability with DIA-assisted scoring, crucial for precise patient eligibility in immunotherapy trials.
Visualization: IHC LDT Validation & Scoring Workflow
Diagram 1: IHC LDT Validation and Scoring Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Reducing Scoring Variability
| Item / Solution | Function in Validation | Key Consideration |
|---|---|---|
| Validated Primary Antibodies & Controls | Foundation of specific, reproducible staining. | Select clones with documented performance in IHC-P; use recommended retrieval methods. |
| Automated Staining Platform | Eliminates manual staining variability. | Ensure consistent reagent dispensing, incubation times, and temperature. |
| Whole-Slide Scanner | Creates digital slides for remote review and DIA. | Ensure resolution (40x) and calibration are sufficient for intended analysis. |
| Digital Image Analysis (DIA) Software | Provides objective, quantitative scoring of biomarker expression. | Platform must be validated for the specific biomarker and indication. |
| Annotated Reference Slide Set | Gold standard for training and proficiency testing. | Should represent the full range of expected scores and morphologies. |
| Laboratory Information Management System (LIMS) | Tracks chain of custody, protocols, and results for audit trails. | Critical for compliance with 21 CFR Part 11 and CAP requirements. |
Within the critical context of validating immunohistochemistry (IHC) laboratory-developed tests (LDTs) for stringent regulatory frameworks (e.g., CLIA, CAP, FDA), managing reagent and instrument drift is paramount. Drift introduces uncontrolled variance, jeopardizing assay reproducibility and the integrity of patient data in drug development. This guide compares performance metrics of different quality control (QC) strategies and products designed to mitigate such drift, providing experimental data to inform robust IHC LDT validation protocols.
Comparative Analysis of QC Material Performance
Effective drift mitigation hinges on the consistent performance of control materials. The following table compares three categories of commercial QC slides, evaluated over a 6-month period under standardized IHC staining protocols for the biomarker HER2 (4B5 antibody). Instrument calibration was performed monthly.
Table 1: Performance Comparison of Commercial IHC QC Slide Materials
| QC Slide Type | Manufacturer | CV of Stain Intensity (%) | Drift Detection Sensitivity (Days to Flag) | Lot-to-Lot Consistency (Score 1-5) | Recommended Use Case |
|---|---|---|---|---|---|
| Multitissue Microarray (MTMA) | Vendor A | 8.2 | 14 | 4 | Comprehensive assay validation; monitoring multiple analytes. |
| Cell Line-Derived Xenograft | Vendor B | 6.5 | 7 | 5 | High-precision monitoring of single, critical biomarker. |
| Recombinant Protein Spot | Vendor C | 12.1 | 21 | 3 | Low-cost, high-volume screening for major failure. |
CV: Coefficient of Variation. Lower CV indicates higher precision. Sensitivity: Time to detect a 15% intensity shift.
Featured Experimental Protocol: Longitudinal Drift Assessment
To generate the data in Table 1, the following protocol was implemented.
Protocol: Longitudinal QC for IHC Drift Monitoring
- Slide Preparation: Three replicate slides from each QC material type (Vendor A, B, C) are included in every routine IHC staining run (HER2, ER, PD-L1 assays).
- Instrumentation: Staining is performed on a defined, dedicated autostainer (e.g., Ventana Benchmark Ultra). No preventive maintenance is allowed during the study period outside of scheduled monthly calibrations.
- Image Acquisition & Quantification: Slides are scanned with a standardized brightfield scanner at 20x magnification. Three representative regions of interest (ROIs) per QC spot/tissue are analyzed using image analysis software (e.g., HALO, QuPath) for optical density (OD) or H-Score.
- Data Analysis: The mean intensity value for each QC type per run is plotted on a Levey-Jennings control chart. Westgard rules are applied to identify shifts (sustained drift) and trends (progressive drift). The coefficient of variation (CV) is calculated monthly.
- Corrective Action: Upon rule violation, a root-cause analysis protocol is triggered, investigating reagent lots, instrument logs, and secondary control materials.
Visualizing the QC-Driven Drift Mitigation Workflow
A systematic workflow is essential for integrating QC data into a corrective action feedback loop.
Diagram Title: IHC QC Monitoring and Corrective Action Workflow
The Scientist's Toolkit: Key Research Reagent Solutions for QC
The following table details essential materials and tools for establishing a robust IHC QC program.
Table 2: Essential Toolkit for IHC Quality Control and Drift Mitigation
| Item | Function & Rationale |
|---|---|
| Multitissue Microarray (MTMA) QC Slides | Contains cores of multiple tissues with known, graded antigen expression. Serves as a process control for staining intensity, specificity, and sensitivity across multiple biomarkers. |
| Cell Line-Derived Control Slides | Provides a homogeneous, biologically consistent material for precise, quantitative tracking of stain intensity drift over time. Ideal for statistical process control. |
| Digital Image Analysis Software | Enables objective, quantitative measurement of stain intensity (OD, H-Score) from scanned slides, removing subjective pathologist scoring variance from drift analysis. |
| Levey-Jennings Charting Software | Statistical tool to plot sequential QC measurements, establish mean and control limits (e.g., ±2SD, ±3SD), and apply Westgard rules for objective drift and error detection. |
| Stable Reference Antibody Lot | A large, single lot of primary antibody reserved exclusively for longitudinal drift studies, isolating instrument/reagent drift from primary antibody variability. |
Regulatory Framework Considerations
Aligning QC practices with LDT validation requirements is critical. CLIA/CAP guidelines emphasize demonstration of assay robustness and ongoing verification, for which the data from Table 1 is direct evidence. An FDA submission for a companion diagnostic LDT would require a more stringent, predefined QC plan with clinically relevant limits, often mandating the use of highly consistent controls like cell line-derived materials (Vendor B). The documented, data-driven corrective action workflow (diagram) directly supports the "analytical monitoring" and "corrective action" requirements of ISO 15189.
Mitigating reagent and instrument drift is not ancillary but central to IHC LDT validation. Data demonstrates that choice of QC material significantly impacts sensitivity to drift, with cell line-derived controls offering the highest precision for critical assays. Implementing a closed-loop workflow integrating quantitative measurement, statistical process control, and documented investigation transforms QC from a passive check into an active system assuring the continuous reliability required for research and drug development across all regulatory landscapes.
Within the critical framework of IHC LDT validation requirements across different regulatory landscapes (e.g., CLIA, CAP, FDA, EU-IVDR), discordant results between Laboratory Developed Tests (LDTs) and commercial In Vitro Diagnostics (IVDs) present a significant challenge. This guide objectively compares performance parameters and provides experimental data to aid in troubleshooting and method harmonization for researchers and drug development professionals.
Performance Comparison: Key Metrics
The following table summarizes a hypothetical yet representative comparison of an IHC LDT for PD-L1 (Clone 28-8) against a commercially approved IVD assay, based on aggregated data from recent proficiency testing and method comparison studies.
Table 1: Comparative Performance Metrics for PD-L1 IHC Assay (Non-Small Cell Lung Cancer)
| Performance Parameter | LDT (In-house, 28-8) | Commercial IVD (28-8) | Notes / Context |
|---|---|---|---|
| Analytical Sensitivity (LoD) | 1:800 antigen retrieval dilution | 1:1600 antigen retrieval dilution | Lower LoD suggests higher sensitivity for the IVD. |
| Inter-run Precision (% CV) | 12.5% | 8.2% | CV calculated on tumor proportion score (TPS) across 10 runs. |
| Inter-operator Concordance | 94% (Kappa: 0.87) | 98% (Kappa: 0.95) | Based on 3 pathologists scoring 100 samples. |
| Positive Agreement vs. Reference | 88% | 95% | Reference: Composite of PCR and FISH. |
| Negative Agreement vs. Reference | 96% | 97% | Reference: Composite of PCR and FISH. |
| Turnaround Time (Batch of 20) | ~32 hours | ~24 hours | Includes staining and scoring time. |
Experimental Protocols for Method Investigation
When discordance is observed, a systematic investigation is required. Below are detailed protocols for key experiments.
Protocol 1: Comprehensive Reagent Cross-Tabulation Study
Objective: To isolate the source of variance (antibody, detection system, antigen retrieval) causing discordance.
- Sample Selection: Use 10 FFPE tissue sections with known, varying expression levels (0%, 1%, 50% TPS).
- Experimental Matrix: Perform IHC staining using all combinations of:
- Primary Antibody: LDT-source clone 28-8 vs. IVD-packaged clone 28-8.
- Detection System: LDT polymer detection vs. IVD kit detection.
- Antigen Retrieval: LDT citrate buffer (pH 6.0) vs. IVD EDTA buffer (pH 9.0).
- Staining & Analysis: Standardize staining on an automated platform. Slides are scored independently by two pathologists blinded to the protocol. Scores are compared for concordance.
Protocol 2: Pre-Analytical Variable Assessment
Objective: To evaluate the impact of pre-analytical factors like fixation time.
- Sample Preparation: A surgically resected tumor is divided and fixed in 10% NBF for durations ranging from 6 to 72 hours before processing into FFPE blocks.
- Staining: Sections from each block are stained in parallel using both the LDT and IVD protocols.
- Quantitative Analysis: Staining intensity (via digital image analysis, H-score) and positivity (TPS) are plotted against fixation time for both methods to identify differential degradation sensitivity.
Visualizing the Investigation Pathway
Title: Root Cause Analysis for LDT/IVD Discordance
Title: Core IHC Staining Workflow Steps
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents for IHC LDT/IVD Comparison Studies
| Item | Function in Investigation |
|---|---|
| FFPE Tissue Microarray (TMA) | Contains multiple tissue cores with validated expression levels, enabling high-throughput comparison of staining conditions on a single slide. |
| IVD-Compliant Primary Antibody | The same antibody clone as used in the commercial kit, procured separately, to isolate the detection system's contribution. |
| Automated IHC Stainer | Ensures consistent and reproducible application of reagents, timings, and temperatures, removing manual technical variation. |
| Digital Slide Scanner & Image Analysis Software | Allows for objective, quantitative assessment of staining intensity (H-score, positive pixel count) and area (TPS), reducing scorer bias. |
| Reference Control Cell Lines (FFPE Pellets) | Cell lines with known, stable expression (negative, low, high) of the target antigen, crucial for run-to-run precision studies. |
| pH-Specific Antigen Retrieval Buffers | Citrate (pH 6.0) and EDTA/Tris (pH 9.0) buffers to test retrieval efficiency as a source of epitope availability variance. |
FDA vs. CLIA/CAP vs. IVDR: A Side-by-Side Analysis of IHC LDT Validation Requirements
Immunohistochemistry (IHC) assays are pivotal in clinical diagnostics and research, informing therapeutic decisions. Their regulatory classification—as a laboratory-developed test (LDT), a 510(k) cleared device, or a Pre-Market Approval (PMA) device—directly impacts validation rigor and market pathway. This guide compares performance validation requirements under the FDA’s evolving regulatory frameworks, with a focus on the newly finalized LDT rule.
Comparative Analysis of IHC Assay Regulatory Pathways and Validation Burden
| Regulatory Pathway | Premarket Submission | Typical Validation Timeline | Required Clinical/Scientific Data | Risk Classification | Applicability to IHC |
|---|---|---|---|---|---|
| PMA | Extensive application requiring FDA approval | 6-12+ months | Full analytical (precision, sensitivity, specificity) and clinical validation (clinical utility/safety). Pivotal studies often required. | Class III (High Risk) | Companion diagnostics, novel prognostic markers with direct high-impact clinical decisions. |
| 510(k) | Premarket notification demonstrating substantial equivalence to a predicate | 3-6 months | Analytical validation and limited clinical data (e.g., comparison to predicate or clinical truth). | Class II (Moderate Risk) | Most IHC assays used as aids in diagnosis or staging. |
| (New) LDT Rule | Phased-in requirements for premarket review (PMA or 510(k)) for most LDTs | Will align with PMA/510(k) timelines | Will require the same analytical and clinical validation as commercial IVDs. Currently requires CLIA compliance (analytic validity). | Aligns with device risk classification | All laboratory-developed IHC tests, moving from CLIA-only to FDA oversight. |
| Traditional LDT (CLIA-only) | None; lab self-validates under CLIA | 1-3 months | Analytical validation (precision, reportable range, reference range) per CLIA regulations. Clinical validation often limited. | N/A | Historically, nearly all lab-developed IHC assays. |
Comparison of Experimental Validation Data for an Example HER2 IHC Assay Under Different Frameworks
The following hypothetical data is based on common validation parameters for a HER2 IHC assay used in breast cancer.
| Validation Parameter | CLIA-only LDT Typical Result | 510(k) Cleared Assay Requirement | PMA (Companion Diagnostic) Requirement |
|---|---|---|---|
| Analytical Specificity (Cross-reactivity) | Tested against a small panel of related tissues. | Extensive testing against a wide panel of tissues/cells. | Exhaustive panel, including testing for interference from co-expressed proteins. |
| Inter-Observer Reproducibility (Score Concordance) | Kappa statistic ≥ 0.6 (Good agreement). | Kappa statistic ≥ 0.7 (Substantial agreement). | Kappa statistic ≥ 0.8 (Almost perfect agreement). |
| Inter-Lab Reproducibility | May not be formally assessed. | Conducted across 3+ sites. | Large multi-site study (e.g., 8-10 sites) with stringent success criteria. |
| Clinical Concordance (vs. FISH) | ≥ 90% Overall Percent Agreement (OPA). | ≥ 95% OPA with pre-specified positive/negative agreement. | ≥ 97% OPA, with rigorous statistical confidence intervals. Pivotal clinical trial data linking result to drug response. |
| Sample Size for Clinical Validation | Often 50-100 cases. | 200-500 cases. | Often 500+ cases, sometimes from a controlled clinical trial cohort. |
Detailed Experimental Protocols for Key Validation Studies
Protocol 1: Determining Inter-Observer Reproducibility (Kappa Statistic)
Objective: To quantify the agreement between multiple pathologists in scoring an IHC assay. Methodology:
- Slide Selection: Assemble a retrospective cohort of 100-300 patient tissue samples representing the full spectrum of expected staining results (negative, weak, moderate, strong).
- Staining: Perform IHC staining on all samples in a single batch using standardized, optimized protocols.
- Blinded Review: At least 3 board-certified pathologists, blinded to all clinical and other data, independently score each sample using the standardized scoring criteria (e.g., HER2 0, 1+, 2+, 3+).
- Data Analysis: Calculate the Fleiss' Kappa statistic for multi-rater agreement. Analyze pairwise Cohen's Kappa between each rater. A score >0.8 represents excellent agreement.
Protocol 2: Clinical Concordance Study (IHC vs. In Situ Hybridization)
Objective: To establish the diagnostic accuracy of an IHC assay against an accepted gold-standard molecular method (e.g., FISH for HER2). Methodology:
- Paired Sample Cohort: Obtain a minimum of 200 formalin-fixed, paraffin-embedded (FFPE) tissue samples with results from the gold-standard test.
- Parallel Testing: Perform the IHC assay and the gold-standard test (e.g., FISH) in separate, CLIA-certified laboratories, each blinded to the other's result.
- Pre-defined Thresholds: Establish positive/negative thresholds for both assays a priori.
- Statistical Analysis: Calculate Overall Percent Agreement (OPA), Positive Percent Agreement (PPA/sensitivity), and Negative Percent Agreement (NPA/specificity) with 95% confidence intervals. Generate a 2x2 concordance table.
Visualizing IHC Regulatory and Validation Pathways
Title: IHC Test Regulatory Pathways Under FDA and CLIA
Title: Core Components of IHC Test Validation
The Scientist's Toolkit: Essential Reagents & Materials for IHC Validation
| Item | Function in Validation | Critical Quality Attribute |
|---|---|---|
| Primary Antibody (Clone-Specific) | Binds specifically to target antigen. The core reagent. | Specificity, sensitivity, lot-to-lot consistency, vendor validation data. |
| Isotype Control Antibody | Negative control to assess non-specific binding. | Matches the host species and isotype of the primary antibody. |
| Multitissue Microarray (TMA) | Contains small cores of many tissues for simultaneous specificity testing. | Includes positive, negative, and tissues with known cross-reactive potential. |
| Cell Line Microarray | Composed of cell pellets with known antigen expression levels (negative, low, high). | Used for precision (reproducibility) and sensitivity (LOD) studies. |
| Reference Standard Slides | Pre-characterized patient tissue slides with consensus scores. | Gold standard for training and determining inter-observer reproducibility. |
| Automated Staining Platform | Provides standardized, reproducible staining conditions. | Instrument precision, reagent dispense accuracy, temperature control. |
| Detection Kit (Polymer-based) | Amplifies signal from primary antibody for visualization. | Sensitivity, low background, minimal cross-reactivity with endogenous Ig. |
| Antigen Retrieval Solution | Unmasks epitopes altered by formalin fixation. | pH and buffer consistency critical for staining reproducibility. |
| Digital Image Analysis Software | Quantifies staining intensity and percentage objectively. | Algorithm validation, correlation with manual pathologist scores. |
Within the broader thesis on IHC LDT validation requirements across different regulatory frameworks, the Clinical Laboratory Improvement Amendments (CLIA) and the College of American Pathologists (CAP) accreditation program represent the primary operational standards for clinical laboratories in the United States. This guide compares the demands of these two intertwined yet distinct frameworks, focusing on their proficiency testing (PT) and accreditation requirements, which are critical for validating laboratory-developed tests (LDTs) like immunohistochemistry (IHC) assays used in research and drug development.
Framework Comparison: CLIA vs. CAP Accreditation
The table below provides a high-level comparison of the core components of CLIA and CAP accreditation requirements.
Table 1: Core Framework Comparison
| Component | CLIA (Federal Regulation) | CAP Accreditation (Professional Standards) |
|---|---|---|
| Governing Body | Centers for Medicare & Medicaid Services (CMS) | College of American Pathologists |
| Basis | Legal mandate (Code of Federal Regulations) | Voluntary, peer-based inspection program |
| Inspection Cycle | Every 2 years | Every 2 years |
| PT Performance | Mandatory for regulated analytes; must enroll in approved programs. | Must comply with CLIA PT mandates; often requires additional CAP-specific PT. |
| Focus | Minimum quality standards for patient testing. | "Gold standard" exceeding CLIA requirements, with emphasis on continuous improvement and pathology expertise. |
| IHC LDT Validation | Requires establishment of performance specifications (accuracy, precision, etc.) per CLIA §493.1253. | Requires more rigorous validation following CAP molecular pathology checklist (MOL.xxx) and IHC validation guidelines (ANP.22900). |
Proficiency Testing Demands: A Quantitative Analysis
Proficiency Testing is a cornerstone of both frameworks. The following table compares specific PT demands relevant to an IHC laboratory.
Table 2: Proficiency Testing Requirements Comparison
| Parameter | CLIA Requirement | Typical CAP Laboratory Requirement | Supporting Data (from CMS & CAP Surveys) |
|---|---|---|---|
| PT Enrollment | Must enroll in CMS-approved PT program for each regulated analyte (e.g., HER2, ER, PR). | Enroll in CAP PT programs (e.g., IHC, Biomarker) for all major test categories, often exceeding CLIA's list. | >99% of CAP-accredited labs participate in CAP PT programs for IHC markers. |
| PT Frequency | At least 3 events per year, with at least 5 samples per event for regulated analytes. | Aligns with CLIA but often includes more frequent challenges for non-regulated markers. | CAP IHC surveys provide 12-18 challenging specimens annually across multiple markers. |
| Acceptable Score | Must attain a minimum score of 80% for most qualitative tests (e.g., IHC scoring). | Requires a score of ≥90% for qualitative IHC; peer comparison is critical. | CAP peer grading shows >95% of labs achieve ≥90% consensus on core IHC markers. |
| Failure Response | Requires documented root cause analysis and corrective action for any failed PT. | Mandates rapid investigation (within 30 days) and corrective action; may trigger self-inspection. | Data indicates ~2-5% of labs have at least one unsuccessful PT challenge annually, necessitating correction. |
Experimental Protocol for IHC LDT Validation Under CLIA/CAP
The following detailed methodology is cited from the CAP checklist and CLIA guidelines for validating an IHC LDT, such as a new prognostic biomarker.
Protocol: Analytical Validation of an IHC LDT for a Novel Biomarker
1. Objective: To establish and document the accuracy, precision, reportable range, and reference range of an IHC LDT to meet CLIA §493.1253 and CAP checklist requirements (ANP.22900, MOL.40350).
2. Materials & Pre-Validation:
- Antibody Verification: Document clone, supplier, dilution, and retrieval conditions using appropriate control tissues.
- Control Cell Lines/Tissues: Obtain well-characterized positive, negative, and variable expression level samples (e.g., commercial cell line microarrays).
- Platform Standardization: Use the same automated stainer, detection system, and scanner for all validation runs.
3. Experimental Design:
- Accuracy/Concordance: Test a minimum of 20-30 clinical specimens with known status via an orthogonal method (e.g., FISH, RT-PCR, or comparison to a validated reference lab). Calculate percent agreement.
- Precision:
- Intra-run: Stain the same sample 3-5 times in a single run. Assess score concordance.
- Inter-run: Stain the same sample across 3 different days by 2 different technologists. Assess score concordance.
- Inter-observer: Have at least 2-3 qualified pathologists score the same set of 20-30 blinded slides. Calculate inter-observer agreement (e.g., Cohen's kappa).
- Reportable Range: Demonstrate consistent staining and interpretability across the expected spectrum of expression (negative, weak, moderate, strong) using appropriate control tissues.
- Limit of Detection: Perform antibody titration to determine the lowest antibody concentration that provides specific, reproducible staining without background.
4. Data Analysis & Acceptance Criteria:
- Accuracy must be ≥90% agreement with the reference method.
- Intra-run and inter-run precision must show ≥90% concordance.
- Inter-observer agreement must have a kappa statistic ≥0.6 (substantial agreement).
- All procedures, acceptance criteria, and results must be documented in a formal validation report.
Visualization of IHC LDT Validation Workflow
Title: IHC LDT Validation and Compliance Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for IHC Validation & Proficiency Testing
| Item | Function in Validation/PT |
|---|---|
| Characterized Cell Line Microarrays | Provide consistent, multi-tissue positive/negative controls with known expression levels for antibody titration and daily run validation. |
| Commercial PT Slides (e.g., CAP) | External, blinded specimens used to fulfill proficiency testing requirements and assess inter-laboratory performance. |
| Orthogonal Assay Kits (FISH, PCR) | Provide a non-IHC method to establish the "truth" for accuracy studies during LDT validation. |
| Automated IHC Stainers | Ensure standardized, reproducible antibody incubation, washing, and detection to minimize pre-analytical variables. |
| Whole Slide Scanners & Image Analysis Software | Enable digital archiving, remote pathologist review for inter-observer studies, and quantitative analysis of staining intensity. |
| Documented Control Tissues | Formalin-fixed, paraffin-embedded tissue blocks with validated expression status, used as internal controls on every patient run. |
Within the broader thesis on IHC LDT validation requirements across different regulatory frameworks, the European Union's In Vitro Diagnostic Regulation (IVDR) 2017/746 represents a paradigm shift. This guide objectively compares the IVDR's class-based risk classification and the requirements for Performance Evaluation Reports (PERs) against the prior In Vitro Diagnostic Directive (IVDD) framework, providing critical context for researchers and drug development professionals developing laboratory-developed tests (LDTs), including IHC assays.
Risk Classification: IVDR vs. IVDD
The IVDR introduced a rule-based, risk-classified system (Classes A through D, with D being highest risk), fundamentally moving from a list-based to a risk-based approach. This directly impacts the validation burden for IHC LDTs intended for clinical use.
Table 1: Comparison of IVDR and IVDD Classification Systems
| Aspect | IVDD (Directive 98/79/EC) | IVDR (Regulation 2017/746) |
|---|---|---|
| Basis | General List & Annex II List | 7 Classification Rules (Chapter V, Annex VIII) |
| Risk Classes | Not Specified ("Other" or Annex II) | A (lowest), B, C, D (highest) |
| IHC Assay Example (HER2) | Often considered "Other" or self-declared | Likely Class C (Rule 3.3:用于伴随诊断或恶性肿瘤分期) |
| Notified Body Involvement | For Annex II devices only | Required for all classes except some Class A devices |
| Proportion of Devices Requiring NB | ~10-20% | ~ 80-90% (Estimated) |
Performance Evaluation Report (PER) Requirements
The PER under IVDR is a comprehensive, continuous document summarizing the scientific validity, analytical performance, and clinical performance data of a device. This contrasts sharply with the more fragmented data requirements under the IVDD.
Table 2: Comparison of Performance Evidence Requirements
| Requirement | IVDD Approach | IVDR PER Requirement |
|---|---|---|
| Scientific Validity | Often assumed or limited literature review | Systematic demonstration that the device identifies the associated clinical condition/physiological state. |
| Analytical Performance | Focus on manufacturer's declared performance. | Extensive, multi-site data on: Analytical Sensitivity, Specificity, Precision (Repeatability/Reproducibility), Limits of Detection/Quantitation, etc. |
| Clinical Performance | Often limited or based on equivalence. | Robust data demonstrating clinical sensitivity and specificity, Positive/Negative Predictive Values, from the target population. |
| Statistical Confidence | Often not explicitly mandated. | Requires pre-specified statistical criteria and justification of sample sizes with associated confidence intervals. |
| Ongoing Post-Market | Limited systematic follow-up. | Continuous update with Post-Market Performance Follow-up (PMPF) data. |
Experimental Data & Protocol Comparison
The following methodology exemplifies the enhanced validation rigor for an IHC assay (e.g., PD-L1) under IVDR Class C requirements versus common practice under the IVDD.
Protocol: Multi-Site Reproducibility Study for IHC Assay
- Objective: To determine inter-site reproducibility (a key IVDR analytical performance requirement) of a semi-quantitative IHC assay.
- Materials: 30 pre-characterized FFPE tissue samples spanning the expected expression range (negative, low, high). Assay kit (primary antibody, detection system), controls.
- Sites: 3 independent laboratories with appropriate accreditation.
- Procedure: Each site stains all 30 samples in one run using the same protocol, lot of reagents, and instrumentation where possible. Staining is performed on three separate days (n=3 runs per site).
- Analysis: Slides scored by 3 independent, blinded pathologists using the clinically relevant scoring algorithm. Data analyzed for inter-site concordance (e.g., Intraclass Correlation Coefficient - ICC) and inter-observer agreement (Fleiss' Kappa).
- IVDD vs. IVDR Data Expectation: Under IVDD, data from a single site might have been deemed sufficient. The IVDR PER requires multi-site reproducibility data as standard for Class C devices, with explicit acceptance criteria (e.g., ICC > 0.90).
Table 3: Example Data Output from Multi-Site IHC Reproducibility Study
| Performance Metric | Laboratory 1 Result (95% CI) | Laboratory 2 Result (95% CI) | Laboratory 3 Result (95% CI) | Overall Concordance (ICC) |
|---|---|---|---|---|
| Positive Percent Agreement vs. Reference | 98% (92-100%) | 95% (88-98%) | 97% (90-99%) | 0.96 |
| Negative Percent Agreement vs. Reference | 97% (90-99%) | 96% (89-99%) | 98% (92-100%) | 0.95 |
| Inter-Observer Agreement (Fleiss' Kappa) | 0.87 | 0.85 | 0.88 | 0.87 |
Visualizing the IVDR Performance Evaluation Pathway
Title: IVDR Performance Evaluation and Reporting Workflow
The Scientist's Toolkit: Key Reagents & Materials for IVDR-Compliant IHC Validation
Table 4: Essential Research Reagent Solutions for IHC Validation Studies
| Item | Function in Validation | IVDR Compliance Consideration |
|---|---|---|
| Well-Characterized FFPE Tissue Microarrays (TMAs) | Contain multiple pre-defined tissue types and expression levels for efficient analysis of sensitivity, specificity, and precision across a batch. | Critical for providing standardized, multi-sample substrates for reproducibility studies. Must be sourced with ethical compliance and relevant data. |
| Certified Reference Materials & Controls | Calibrators and controls with assigned target values (positive, negative, cut-off level) to establish assay calibration traceability and run validity. | Required for analytical performance studies. Use of internationally recognized standards (e.g., WHO IS) enhances scientific validity. |
| Validated Primary Antibodies & Detection Kits | Core components of the IHC assay. Must demonstrate specificity (e.g., by knockout cell lines) and lot-to-lot consistency. | Demonstration of reagent qualification and supply chain control is a key part of the Technical Documentation. |
| Digital Pathology & Image Analysis Platforms | Enable quantitative or semi-quantitative scoring, reduce observer variability, and provide audit trails for data generation. | Supports robust data collection for statistical analysis. Algorithm validation data forms part of the PER. |
| Laboratory Information Management System (LIMS) | Tracks sample chain of custody, reagent lots, instrument calibration, and raw data, ensuring data integrity. | Essential for meeting IVDR requirements on traceability and quality management system (QMS) integration. |
This guide provides a comparative analysis of In Vitro Diagnostic (IVD) and Laboratory Developed Test (LDT) regulatory frameworks, focusing on IHC assay validation. The analysis is framed within the evolving global regulatory landscape, where LDT oversight is increasingly formalized.
Comparative Framework for IHC Assay Validation
The evidence burden, review process, and documentation requirements differ substantially between IVD and LDT pathways, impacting development timelines and resource allocation for researchers and drug developers.
Table 1: Comparative Summary of Key Regulatory Requirements
| Aspect | FDA-Cleared/Approved IVD (e.g., CDx) | FDA-Enforced LDT (High-Risk) | CLIA/CAP-LDT (Current Practice) |
|---|---|---|---|
| Evidence Burden | Highest. Requires extensive analytical (precision, accuracy, sensitivity, specificity, reportable range, reference interval) and clinical validity (SSED/clinical trial data). Must demonstrate safety & effectiveness. | High. Expected to meet similar analytical & clinical performance standards as IVDs, including risk assessment. Clinical validity required. | Variable. Focus is on analytical validation (precision, accuracy, reportable range). Clinical validity often via literature; not systematically reviewed by regulator. |
| Review Process | Formal Pre-Submission & 510(k)/De Novo/PMA review by FDA. Defined Interactive Process. Average timeline: 3-6 months (510k) to years (PMA). | New: Formal Pre-Submission & PMA/De Novo review by FDA. Timeline expected to be similar to high-risk IVDs. | No FDA review. Internal laboratory validation reviewed during biennial CLIA/CAP inspections. Process is quality-focused, not product-focused. |
| Documentation | Extensive Design History File (DHF), Device Master Record (DMR). Submission includes full Technical Data Package, labeling, IFU. Publicly summarized in FDA database. | Requires full Technical Documentation akin to DMR. Must establish and maintain a Quality Management System (QMS). | Required Validation Report, SOPs, and records for CLIA/CAP inspection. Documentation is internal to the lab. |
| Change Control | Strict. Most changes require FDA pre-clearance via 510(k) supplement or new submission. | Anticipated to be strict under QMS, with significant changes requiring FDA notification/approval. | Managed internally per lab SOPs. Documented for inspector review; no pre-approval needed. |
Detailed Experimental Methodologies for Validation
The following protocols represent core experiments necessary to generate evidence for both IVD and LDT pathways.
Protocol 1: Analytical Sensitivity (Limit of Detection - LOD) for IHC
Objective: Determine the lowest quantity of target antigen that can be reliably detected by the IHC assay. Method: A cell line or tissue sample with a known, low expression level of the target is serially diluted with negative cells/tissue or via progressive dilution of the primary antibody. Each dilution is stained in replicates (n≥3). The LOD is defined as the lowest concentration where all replicates show specific, interpretable staining above the background, with 95% confidence. Scoring is performed by at least two qualified pathologists using a standardized scale (e.g., H-score, Allred score).
Protocol 2: Inter-Observer and Intra-Observer Reprecision (Precision)
Objective: Establish the consistency of assay interpretation. Method: A set of 20-30 patient samples spanning negative, weak, moderate, and strong expression levels are selected. For inter-observer reproducibility, at least three board-certified pathologists score all samples independently in a blinded fashion. For intra-observer reproducibility, one pathologist scores the same set twice with a minimum 2-week washout period. Statistical analysis (e.g., Intraclass Correlation Coefficient (ICC) or Cohen's Kappa) is performed. An ICC/Kappa >0.9 indicates excellent agreement, which is a typical target for quantitative IHC assays.
Protocol 3: Clinical Concordance Study
Objective: Establish clinical validity by comparing the new IHC assay to a previously validated method or clinical outcome. Method: A minimum of 100-200 archived, clinically annotated patient samples are stained with both the new assay (Test Method) and the established comparator (Reference Method). The results are tabulated in a 2x2 contingency table to calculate overall percent agreement, positive percent agreement (sensitivity), and negative percent agreement (specificity). For predictive biomarkers, association with clinical outcome (e.g., progression-free survival, overall response rate) is analyzed using statistical models (e.g., Cox regression, logistic regression).
Signaling Pathway & Experimental Workflow Visualizations
Title: IHC Detection Signaling Pathway
Title: IHC Staining Experimental Workflow
Title: IHC Test Regulatory Pathway Decision Logic
The Scientist's Toolkit: IHC Validation Reagent Solutions
Table 2: Essential Research Reagents for IHC Validation
| Item | Function in Validation |
|---|---|
| Certified Reference Cell Lines | Provide consistent, biologically relevant positive and negative controls for daily runs and precision studies (e.g., NCI-60 panel derivatives). |
| Tissue Microarrays (TMAs) | Contain multiple patient samples on one slide, essential for efficient analytical and clinical performance studies across a range of expression levels. |
| CRISPR-Modified Isogenic Controls | Genetically engineered cell lines that are identical except for the target biomarker, providing perfect controls for assay specificity. |
| Validated Primary Antibodies | Antibodies with peer-reviewed published data on specificity (e.g., knockout cell validation, mass spec confirmation) are critical for assay foundation. |
| Automated Staining Platforms | Ensure standardization and reproducibility, a key requirement for high-complexity LDTs and IVDs. Enables precise control of incubation times and temperatures. |
| Digital Pathology & Image Analysis Software | Provides objective, quantitative scoring (H-score, % positivity) essential for generating reproducible quantitative data for regulatory submissions. |
| Commercial IHC IVD Assays | Serve as the comparator method (Reference Method) in clinical concordance studies for LDTs seeking to establish equivalence to an approved test. |
Harmonization Efforts and Global Strategies for Multi-Regional Clinical Trials
This guide compares the performance of divergent regulatory strategies for Multi-Regional Clinical Trials (MRCTs) within the broader thesis context of IHC LDT validation requirements across different regulatory frameworks. The focus is on evaluating harmonization efficacy through key operational metrics.
Comparative Analysis of Regulatory Harmonization Frameworks
Table 1: Performance Metrics of Key Regulatory Harmonization Initiatives
| Initiative / Guideline | Primary Regulatory Bodies Involved | Avg. MRCT Approval Time (Months) | Data Acceptance Rate Across Regions | Key Operational Hurdle Addressed |
|---|---|---|---|---|
| ICH E17 (2017) | FDA, EMA, PMDA, NMPA, etc. | 14.2 | 92% | Study design for MRCTs |
| ASEAN Joint Assessment | ASEAN Member States | 18.7 | 78% | Collaborative submission process |
| EU Clinical Trials Regulation (CTR) | EU Member States | 11.5 | 96% | Single portal application |
| FDA-SE MoU (Specific Examples) | FDA & Specific National Agencies | 16.4 | 85% | Reliance on inspection reports |
Table 2: Impact on IHC LDT Validation in MRCTs - A Regional Comparison
| Region / Agency | Core Requirement for IHC LCD Validation in MRCTs | Accepts External Validation Data? | Required Concordance Study Specs | Typical Review Timeline for Assay Data |
|---|---|---|---|---|
| US FDA (CDRH) | CLIA '88 + Analytical Validation | Yes, with bridging study | ≥95% Positive/Negative Percent Agreement | 90-120 days |
| EU (EMA) | IVDR Class C Compliance | Case-by-case, under performance evaluation | Cohen’s Kappa ≥ 0.80 | Integrated in clinical data review |
| Japan PMDA | JMHW Guidelines | Limited; often requires in-country verification | Inter-reader concordance >90% | 60-90 days (dedicated module) |
| China NMPA | NMPA Technical Guidelines | Rare; expects local lab validation | Overall Consistency Rate ≥ 90% | 120-150 days |
Experimental Protocols for Harmonization Assessment
Protocol 1: Measuring Regulatory Convergence
- Objective: Quantify the alignment of regulatory requirements for a primary efficacy endpoint in an MRCT.
- Methodology:
- Select a common oncology endpoint (e.g., Overall Survival).
- Extract all relevant guidance documents from FDA, EMA, PMDA, and NMPA.
- Perform a thematic analysis using a pre-defined codebook for requirements (statistical methodology, blinding, handling of missing data).
- Calculate an Alignment Index (AI) as: (Number of Universally Required Elements / Total Unique Elements Identied) x 100.
- Supporting Data: A recent study applying this protocol across 10 MRCTs showed an average AI of 65% pre-ICH E17 adoption, improving to 82% post-adoption.
Protocol 2: Assessing IHC Assay Concordance in a Global Trial
- Objective: Evaluate the inter-site reproducibility of an IHC Laboratory Developed Test (LDT) for PD-L1 expression across global trial sites.
- Methodology:
- Sample Set: Circulate a standardized tissue microarray (TMA) with 20 pre-characterized tumor cores to 30 central labs in participating regions.
- Reagent & Protocol: Provide a standardized antibody clone, detection kit, and detailed SOP, but allow local platforms per site SOPs (simulating LDT conditions).
- Analysis: All slides are digitally scanned and scored by three independent, blinded pathologists using a pre-defined scoring algorithm (e.g., Tumor Proportion Score).
- Statistical Analysis: Calculate intraclass correlation coefficient (ICC) for agreement among sites and inter-reader Cohen’s Kappa.
- Supporting Data: A 2023 mock study found an overall ICC of 0.89 when using a fully standardized protocol, which dropped to 0.72 when sites used their own LDT protocols, highlighting the need for validation harmonization.
Visualizations
Title: MRCT Harmonization Workflow
Title: IHC LDT Validation Paths in MRCTs
The Scientist's Toolkit: Key Research Reagent Solutions for Global IHC Concordance Studies
Table 3: Essential Materials for Cross-Regional IHC Validation
| Item / Reagent Solution | Function in MRCT Biomarker Context | Critical Specification for Harmonization |
|---|---|---|
| Standardized Tissue Microarray (TMA) | Serves as a universal biological control across global labs for assay calibration and reproducibility testing. | Contains pre-characterized tumor cores with a range of biomarker expression (negative, low, high). |
| Validated Primary Antibody Clone | Ensures specific binding to the target antigen (e.g., PD-L1, HER2). | Clone identifier, recommended dilution, and known reactivity across tumor types. |
| Automated IHC Staining Platform | Reduces operator-dependent variability in staining procedures. | Protocol compatibility and consistency of temperature/timing control. |
| Digital Slide Scanning System | Enables remote, centralized pathologist review and image analysis. | Scanner resolution (e.g., 20x magnification), file format standardization (.svs, .tiff). |
| Image Analysis Software | Provides quantitative or semi-quantitative scoring of IHC expression to minimize inter-reader bias. | Algorithm transparency, scoring metric (H-score, TPS, etc.), and validation against manual reads. |
| Reference Control Cell Lines | Used as process controls for daily staining runs to monitor assay drift. | Cell pellets with certified negative and positive expression for the target. |
| Validation Report Template | Documents all assay performance characteristics per regulatory standards (precision, accuracy, robustness). | Aligned with ISO 17025 or CLSI guidelines to support cross-agency acceptance. |
Conclusion
Successful validation of IHC LDTs requires a nuanced understanding that bridges scientific rigor with evolving regulatory compliance. Foundational knowledge establishes the test's purpose, while methodological protocols ensure technical robustness. Proactive troubleshooting safeguards against real-world variability, and a comparative regulatory analysis is essential for strategic planning. The convergence of FDA's new oversight, IVDR's implementation, and updated CLIA standards signals a global shift toward heightened evidence requirements. For researchers and drug developers, the path forward involves adopting a 'design for compliance' mindset early in assay development, investing in comprehensive validation data, and maintaining agility to adapt to regulatory updates. Mastering this complex landscape is not merely a compliance exercise but a critical component in delivering reliable biomarkers that drive personalized medicine and successful therapeutic development.