Navigating the IVD Regulatory Divide: A Strategic Comparison of US FDA and EU IVDR Requirements
This article provides a comprehensive analysis for researchers, scientists, and drug development professionals on the evolving regulatory landscapes for In Vitro Diagnostics (IVDs) in the United States and European Union.
Navigating the IVD Regulatory Divide: A Strategic Comparison of US FDA and EU IVDR Requirements
Abstract
This article provides a comprehensive analysis for researchers, scientists, and drug development professionals on the evolving regulatory landscapes for In Vitro Diagnostics (IVDs) in the United States and European Union. It explores the foundational principles of the US FDA's pro-innovation framework and the EU's IVDR precautionary approach, detailing practical pathways to market, strategies to overcome current challenges like notified body bottlenecks and LDT rule changes, and a comparative validation for global product strategy. The analysis synthesizes key takeaways to guide strategic decision-making for successful and efficient global market entry.
Understanding the US and EU IVD Regulatory Philosophies and Frameworks
In the global landscape for in vitro diagnostics (IVDs), the regulatory philosophies of the United States and the European Union have significantly diverged. The U.S. Food and Drug Administration (FDA) has cultivated a pro-innovation stance, exemplified by its 510(k) premarket notification pathway. This approach emphasizes efficient market access for new devices by leveraging existing knowledge and predicates [1] [2]. In contrast, the European Union’s framework under the In Vitro Diagnostic Regulation (IVDR) prioritizes a precautionary principle, resulting in a more complex, evidence-intensive, and time-consuming conformity assessment process [1] [3]. This comparison guide, framed within broader research on US-EU regulatory requirements, objectively analyzes the performance of these two pathways for IVD manufacturers, providing critical data for researchers, scientists, and development professionals strategizing global market access.
Comparative Analysis of Regulatory Frameworks
The foundational structures governing IVDs in the US and EU are built on differing principles, directly impacting innovation cycles and market strategy.
Table 1: Foundational Comparison of US and EU IVD Regulatory Systems
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Principle | Pro-innovation, risk-based with predicate reliance [1] [2] | Precautionary, safety-focused with high evidence thresholds [1] [3] |
| Legal Basis | Federal Food, Drug & Cosmetic Act; 21 CFR Parts 809, 812, 820 [3] | Regulation (EU) 2017/746 (IVDR) [3] |
| Risk Classification | 3 Classes (I, II, III) [3] | 4 Classes (A, B, C, D) [3] |
| Primary Market Entry Pathway | Premarket Notification 510(k) (for Class II majority) [4] | Conformity Assessment via a Notified Body (for Classes B, C, D) [3] |
| Core Market Entry Concept | Substantial Equivalence (SE) to a predicate device [4] | Conformity to General Safety and Performance Requirements (GSPRs) [3] |
| "Grandfathering" Provision | Yes (for pre-amendment 1976 devices and cleared predicates) [4] [3] | No. All devices must comply with IVDR; legacy IVDD devices require transition [3] |
| Key Strategic Impact | Enables iterative innovation and faster time-to-market for incremental advances [1] [5]. | Creates a higher initial barrier, potentially slowing initial market entry but aiming for high safety assurance [1] [6]. |
Comparison of Market Entry Pathways and Performance
The operational experience of navigating the FDA 510(k) and EU IVDR pathways reveals stark contrasts in process, duration, and resource allocation.
Table 2: Performance Comparison of Primary Market Entry Pathways
| Metric | FDA 510(k) Pathway | EU IVDR Conformity Assessment |
|---|---|---|
| Typical Timeline to Authorization | 6-12 months from submission to clearance [5] [6]. | 12-18 months or more for Notified Body review and certification [1] [6]. |
| Formal Pre-Submission Feedback | Available via Q-Submission process [7] [5]. | No official pre-submission; limited to scientific advice from authorities or informal NB consultation [7] [5]. |
| Review Structure | Centralized review by the FDA with a statutory 90-day goal; often involves Additional Information cycles [4] [2]. | Decentralized review by a chosen Notified Body (NB). No legally binding review deadline, leading to variability [1] [6]. |
| Documentation Volume | Focused submission; typically hundreds of pages centered on SE demonstration [5]. | Extensive Technical Documentation (thousands of pages) covering full device lifecycle per Annexes II & III of IVDR [5] [6]. |
| Typical Cost (Excluding Clinical Studies) | Estimated $1M - $6M, influenced by complexity [6]. | Estimated $500K - $2M for NB fees and compliance work, influenced by NB and device class [6]. |
| First-to-Market Strategy | The preferred first launch market for 40% more large IVD makers since IVDR [1]. | First launch choice has declined due to complexity and longer timelines [1]. |
| Facility Inspection | Not typically required pre-clearance; QSR inspection can occur anytime post-market [4]. | Audit of manufacturer's Quality Management System (per ISO 13485) is mandatory part of NB assessment for most classes [3] [6]. |
Visualization: The 510(k) Substantial Equivalence Decision Pathway The core of the 510(k) pathway is a logical determination of Substantial Equivalence, as defined by the FDA [4].
Diagram 1: The FDA 510(k) Substantial Equivalence Decision Logic (100 characters)
Visualization: Comparative US vs. EU IVD Regulatory Workflow The high-level workflows for market authorization illustrate fundamental process differences [1] [4] [3].
Diagram 2: Comparative US FDA 510(k) and EU IVDR Market Access Workflows (100 characters)
Detailed Experimental Protocols & Evidence Requirements
A critical divergence lies in the nature and extent of performance evidence required. The 510(k) pathway often relies on well-established bench testing protocols against a predicate, while the IVDR mandates a comprehensive Performance Evaluation with stronger clinical components [3] [7].
Protocol for Analytical Performance Validation (Common to Both Regions)
This protocol forms the basis for demonstrating an IVD's technical capability, essential for both FDA 510(k) and EU IVDR submissions.
- Objective: To establish and validate the key analytical performance characteristics of a new quantitative IVD assay.
- Methodology:
- Precision Testing: Conduct a 20-day experiment following CLSI EP05-A3 guidelines. Test two levels of controls and three human serum pools (low, mid, high analyte concentration) in duplicate, across two runs per day. Calculate within-run, between-run, and total coefficient of variation (%CV) [7].
- Linearity and Reportable Range: Prepare a series of at least 5 dilutions spanning the claimed measuring range from a high-concentration sample. Each dilution is analyzed in triplicate. Perform polynomial regression; the assay is linear if a 1st-order model fits and the deviation from linearity is less than the allowable specification (e.g., ≤10%) [7].
- Limit of Blank/Detection/Quantitation (LoB/LoD/LoQ): Analyze at least 20 replicates of a blank matrix (LoB). LoD is estimated as LoB + 1.645(SD of low-level sample). LoQ is determined by testing samples at low concentrations and identifying the lowest level with a total %CV ≤20% and bias within ±20% [7].
- Method Comparison: Analyze approximately 40-100 clinical samples spanning the assay range using both the new test and a validated comparator method (predicate device or reference method). Perform correlation (e.g., Passing-Bablok regression) and bias analysis (Bland-Altman plot) [7].
Protocol for Clinical Performance Study (Divergent Regional Emphasis)
While both regions require clinical evidence, its generation and role differ significantly.
- Objective (FDA 510(k)): To demonstrate the new device is as safe and effective as the predicate, primarily when non-clinical testing is insufficient to establish Substantial Equivalence for differences in technological characteristics [7] [2].
- Objective (EU IVDR): To establish and confirm the device's scientific validity, analytical and clinical performance, and to fulfill the ongoing requirements of the Performance Evaluation Report [3] [7].
- Methodology & Key Divergences:
- Study Design: Both may use prospective or retrospective sample collections. The FDA often accepts well-designed retrospective studies using archived specimens [7]. The IVDR emphasizes the suitability of specimens for the claimed intended use population.
- Comparator: For the FDA, the primary comparator is often the predicate device [7]. For the IVDR, the comparator may be a clinical standard of truth (e.g., a diagnostic standard or another accepted method), not necessarily another IVD [3].
- Sample Size Justification: The FDA focuses on statistical confidence for claims (e.g., sensitivity, specificity) against the predicate [7]. The IVDR requires a justification based on the device's risk class, intended use, and literature, with an expectation for larger sample sizes for higher classes to ensure representativeness [3].
- Endpoint Analysis: The FDA review centers on the performance claims (e.g., non-inferiority margins). The IVDR requires integration of results into a broader Performance Evaluation Report, linking analytical and clinical data to the device's scientific validity and the General Safety and Performance Requirements [3].
Table 3: Comparison of Clinical Evidence Requirements
| Requirement | FDA 510(k) Pathway | EU IVDR Pathway |
|---|---|---|
| Clinical Evidence Mandate | Not always required. Driven by the need to address differences from the predicate [7] [2]. | Mandatory for all classes. Part of the continuous Performance Evaluation [3]. |
| Acceptable Evidence Sources | Prospective clinical trial, retrospective sample study, literature (sometimes) [7]. | Clinical investigation, study using equivalent device (under strict conditions), literature review [3] [6]. |
| Equivalence Claims | Core of the pathway: "Substantial Equivalence" to a predicate [4]. | Extremely restrictive: Requires access to predicate's technical and clinical data, and same intended purpose, design, and biological interaction [3] [6]. |
| Post-Market Evidence | Collected via post-market surveillance and registries; may be used to support new indications. | Formally required as Post-Market Performance Follow-up (PMPF), which actively updates the Performance Evaluation [3]. |
The Scientist's Toolkit: Key Reagents & Materials for IVD Validation
Successful regulatory submission relies on high-quality, well-characterized materials. Below is a toolkit of essential research reagent solutions.
Table 4: Essential Research Reagent Solutions for IVD Performance Studies
| Reagent/Material | Function in Validation Studies | Critical Quality Attributes |
|---|---|---|
| Certified Reference Material (CRM) | Serves as the primary standard for assigning values to calibrators. Establishes traceability to a higher-order reference method or system (e.g., WHO International Standard). | Purity, commutability (behaves like patient sample), stability, value assignment with stated uncertainty [7]. |
| Clinical Specimen Panels | Used for precision, linearity, interference, method comparison, and clinical sensitivity/specificity studies. Represents the real-world matrix (e.g., human serum, plasma, whole blood). | Well-characterized disease state/analyte concentration, informed consent, integrity (no freeze-thaw degradation), relevant clinical metadata [7]. |
| Stable Control Materials | Monitored daily to verify the assay's performance within established ranges (precision, trend analysis). Used throughout the 510(k) and IVDR stability studies. | Homogeneity, long-term stability, matrix matching to patient samples, target values covering key medical decision points [7]. |
| Interference Test Substances | Systematically added to samples to evaluate assay susceptibility to common interferents (e.g., hemoglobin, bilirubin, lipids, common drugs). | High purity, prepared at supraphysiological concentrations to demonstrate assay robustness [7]. |
| Cell Lines or Microbial Stocks | For infectious disease tests, provide standardized targets for Limit of Detection (LoD) studies and inclusivity/exclusivity testing. | Authenticated identity, standardized titer or concentration, genetic characterization relevant to claimed targets [7]. |
| Biopreservation Medium | Ensures viability and stability of cellular or microbial analytes in clinical specimens from collection through testing, critical for study integrity. | Validated for compatibility with the assay, maintains analyte stability for the duration of the pre-test storage period [7]. |
The strategic impact of these differing pathways is quantifiable in market behavior, timing, and resource allocation.
Table 5: Quantitative Outcomes of the Diverging Regulatory Pathways (2025 Data)
| Outcome Metric | US Market (FDA 510(k)) | EU Market (IVDR) | Implication |
|---|---|---|---|
| Global Market Share | ~46.4% of global MedTech market [1] | ~26.4% of global MedTech market [1] | The US is a larger, more dominant first-tier market. |
| Change in First Launch Preference | Increased as first launch market [1] | Decreased by ~40% for large IVD makers since IVDR [1] | Clear industry shift towards "US-First" launch strategies. |
| Review Timeline Predictability | High (90-day FDA goal, though often extended) [4] [6] | Low (Notified Body reviews average 13-18 months, no legal deadline) [1] [6] | US offers more predictable project planning. |
| System Capacity | Centralized FDA review. | Constrained (51 Notified Bodies for ~28,489 MDR applications) [1]. | EU bottlenecks cause significant delays. |
| Modernization for AI/ML | Predetermined Change Control Plan (PCCP) allows pre-approved, iterative updates [1]. | Dual regulation under IVDR and the EU AI Act; more rigid [1]. | US framework is more adaptive to software-driven innovation. |
Strategic Implications for IVD Developers
The data supports a clear strategic imperative: the FDA's 510(k) pathway, reinforced by the Safety and Performance Based route and modern digital submission tools (eSTAR), offers a faster, more predictable, and pro-innovation route to initial market access [8] [2]. This has solidified the "US-First" launch model, allowing companies to generate early revenue and real-world evidence [1].
Conversely, the EU IVDR presents a higher initial barrier but grants access to a consolidated market of 27 nations. Its emphasis on rigorous lifecycle management, including Post-Market Performance Follow-up (PMPF) and Periodic Safety Update Reports (PSURs), mandates a different, more sustained evidence-generation strategy [3]. Notably, the European Commission has recognized systemic challenges, launching a "Call for Evidence" in late 2025 to simplify the MDR/IVDR, indicating potential future recalibration [1] [9].
For researchers and developers, the optimal global strategy involves sequential market entry: leveraging the efficient 510(k) for initial US commercialization, while concurrently preparing the more comprehensive documentation required for the EU IVDR. Mastery of both frameworks is not merely a regulatory task, but a core component of competitive product development and global commercial strategy.
The introduction of the European Union's In Vitro Diagnostic Regulation (IVDR) represents a paradigm shift in the regulatory landscape, fundamentally moving away from the principles of the former In Vitro Diagnostic Directive (IVDD). Framed within a broader thesis comparing US and EU regulatory requirements for in vitro diagnostics (IVDs), this transition is characterized by a more stringent application of the precautionary principle. This principle mandates proactive risk management and demands a higher standard of clinical and analytical evidence to ensure patient safety and device performance before market entry. The following comparison guides objectively analyze the performance of the current EU system (IVDR) against its predecessor (IVDD) and the parallel US framework, supported by structured data on regulatory outcomes and detailed methodological requirements.
Comparative Analysis of US (FDA) and EU (IVDR) Regulatory Frameworks
The regulatory philosophies of the United States and the European Union, while both risk-based, manifest in distinctly different structures, pathways, and evidential requirements for IVDs.
Risk Classification Systems: A Structural Comparison
The foundation of both systems is risk classification, which dictates the rigor of the pre-market review. The US FDA employs a three-class system (I, II, III), whereas the EU IVDR utilizes a four-class system (A, B, C, D), with Class D representing the highest risk [3]. This difference significantly impacts the volume of devices requiring third-party review.
Table 1: Comparison of US FDA and EU IVDR Risk Classification and Pathways
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Legal Basis | Food, Drug & Cosmetic Act; 21 CFR Parts 809, 812, 814 [10]. | Regulation (EU) 2017/746 [11]. |
| Risk Classes | Class I (Low), Class II (Moderate), Class III (High) [3]. | Class A (Low), Class B, Class C, Class D (High) [3]. |
| Classification Driver | Intended use and predicate comparison [12]. | Rules-based (Annex VIII of IVDR), considering public/patient risk [3]. |
| Key Premarket Pathway (High-Volume) | 510(k) (Demonstration of Substantial Equivalence to a predicate) [10]. | Conformity Assessment via Notified Body (Requires Performance Evaluation Report) [3]. |
| Key Premarket Pathway (Novel/High-Risk) | De Novo (novel, low-moderate risk) or Premarket Approval (PMA) (Class III) [10]. | Conformity Assessment via Notified Body with stricter clinical evidence (Annexes IX-XI) [3]. |
| Clinical Evidence Requirement | For 510(k): Often analytical performance vs. predicate; clinical studies sometimes needed [10]. For PMA: Clinical investigations required [10]. | Performance Evaluation mandatory for all classes: Analytical & Clinical Performance + Scientific Validity [3]. |
| Accelerated Pathway | Breakthrough Devices Program (BDP) [13]. | No formal accelerated pathway; reliance on expert panels and EU reference labs [13]. |
| Post-Market Surveillance (PMS) | Adverse event reporting (MAUDE), periodic reports for PMA [3]. | Proactive, continuous PMS. PSUR required for Class C & D; PMSR for Class A & B [3]. |
| Unique System Requirements | CLIA '88 categorization (Waived, Moderate, High Complexity) [10]. | General Safety & Performance Requirements (GSPR), EUDAMED registration [3]. |
A critical divergence is in the scope of oversight. Under the previous IVDD, an estimated 10-20% of IVDs required Notified Body review. The IVDR has inverted this, subjecting approximately 80-90% of devices to such scrutiny due to its rules-based classification [3]. This expansion is a direct application of the precautionary principle, minimizing reliance on manufacturer self-declaration.
Market Authorization Pathways and Timelines
The regulatory journey from development to market differs substantially. The US system offers more defined, centralized pathways with established timelines. The FDA's Pre-Submission process allows for early, binding feedback [10], and programs like the Breakthrough Devices Program (BDP) provide prioritized interaction. Data shows BDP-designated devices receive marketing authorization significantly faster (e.g., 230 days for PMA vs. 399 days standard) [13].
The EU pathway is decentralized, relying on conformity assessment by independent Notified Bodies. While the IVDR mandates stricter evidence, it lacks a formal accelerated pathway [13]. The process duration can be less predictable, influenced by Notified Body capacity and the complexity of the Performance Evaluation. This structural difference means that for novel, high-risk devices, the US BDP may offer a more predictable and faster route to initial market access, whereas the EU system embeds a higher evidence threshold from the outset.
The Evidence Benchmark: Clinical Performance and Performance Evaluation
The most profound shift from IVDD to IVDR is in the requirement for clinical evidence. The IVDR mandates a continuous, life-cycle based Performance Evaluation (PE) consisting of three pillars: scientific validity, analytical performance, and clinical performance [3].
Table 2: Comparison of Clinical Evidence Requirements
| Requirement | US FDA (Typical 510(k) Pathway) | EU IVDD (Legacy System) | EU IVDR (Current System) |
|---|---|---|---|
| Core Principle | Substantial Equivalence to a predicate device [10]. | Self-declaration of conformity to Essential Requirements for majority of devices. | Demonstration of safety, performance, and scientific validity per General Safety & Performance Requirements (GSPRs) [3]. |
| Primary Data Focus | Analytical performance (accuracy, precision) compared to a predicate; clinical samples may be used [10]. | Varied; often limited technical performance data. | Tripartite Performance Evaluation: Analytical Performance, Clinical Performance, and Scientific Validity [3]. |
| Clinical Performance Study Need | Not routinely required for 510(k); requested if analytical link to clinical outcome is unclear [10]. | Rarely required for non-List A/B devices. | Required where not waived, proportional to device class and novelty. Post-Market Performance Follow-up (PMPF) plans are mandatory [3]. |
| Equivalence Route | Central to the 510(k) pathway [10]. | Commonly used. | Severely restricted under IVDR. Can only be claimed under strict conditions (same manufacturer or legal agreement, same technical/scientific characteristics) [14]. |
This table highlights the IVDR's departure from a predicate-based model. The IVDR's restrictions on claiming equivalence mean manufacturers must often generate new clinical performance data for their specific device, even if similar products exist [14]. This aligns with the precautionary principle by ensuring claims are validated by direct evidence rather than inference.
Experimental Protocols for Regulatory Performance Evaluation
Generating evidence for IVDR compliance requires rigorous, predefined experimental methodologies. Below are generalized protocols for key studies.
Protocol for Analytical Performance Studies
Objective: To determine the analytical sensitivity (detection limit), analytical specificity (interference), accuracy, and precision of the IVD device. Methodology:
- Sample Preparation: Use well-characterized clinical samples or validated reference materials. For detection limit, prepare serial dilutions of the target analyte in a relevant matrix.
- Testing Protocol: Run replicates (n≥20) across multiple days, operators, and lots of reagents (if applicable) following the device's instructions for use.
- Data Analysis:
- Accuracy/Bias: Compare mean results to a reference method or assigned value. Calculate percent bias.
- Precision: Calculate within-run, between-run, and total coefficient of variation (CV%).
- Analytical Sensitivity (LoD): Determine the lowest concentration detected in ≥95% of replicates.
- Analytical Specificity: Test cross-reactivity with structurally similar compounds and interference from common endogenous substances (e.g., hemoglobin, lipids).
Protocol for Clinical Performance Studies
Objective: To establish the clinical sensitivity and specificity (or positive/negative percent agreement) of the IVD device in the target population. Methodology:
- Study Design: Prospective, retrospective, or diagnostic case-control study, as appropriate. Define clear inclusion/exclusion criteria.
- Comparator Method: Use a clinically accepted reference standard (e.g., clinical diagnosis, FDA-approved/CE-marked comparator, well-established laboratory method).
- Sample Size Calculation: Based on pre-specified confidence intervals for sensitivity and specificity (e.g., 95% CI width).
- Testing & Blinding: Test all enrolled samples with the investigational device and the comparator method. Use blinding to prevent interpretive bias.
- Statistical Analysis: Construct a 2x2 contingency table. Calculate clinical sensitivity, specificity, and their confidence intervals. For quantitative assays, perform correlation and Bland-Altman analysis against the comparator.
Protocol for Stability Studies
Objective: To establish the shelf-life (in-use and real-time stability) of reagents and the device. Methodology:
- Real-Time Stability: Store device/reagents under labeled conditions. Test performance at defined intervals (e.g., 0, 3, 6, 9, 12, 18, 24 months) using the analytical performance protocol.
- In-Use/Post-Opening Stability: After opening or reconstitution, test the reagents at defined intervals over the claimed in-use period under simulated use conditions.
- Accelerated Stability: Store products at elevated stress conditions (e.g., temperature) to predict degradation rates and support tentative shelf-life claims during development.
Visualizing Regulatory Pathways and Logic
The following diagrams clarify the logical flow of IVDR classification and contrast the high-level approval pathways between the US and EU systems.
Diagram Title: IVDR Risk Classification Rule Logic Flow (Simplified)
Diagram Title: US vs EU IVD Regulatory Approval Pathway Comparison
The Scientist's Toolkit: Essential Reagents & Materials for IVD Development and Validation
Successful navigation of the IVDR's stringent requirements depends on high-quality, well-characterized materials. This toolkit details essential components for performance studies.
Table 3: Key Research Reagent Solutions for IVD Performance Evaluation
| Reagent/Material | Primary Function in Regulatory Studies | Key IVDR Consideration & Application |
|---|---|---|
| Certified Reference Materials (CRMs) | Provide an absolute standard with defined purity and concentration for calibrating assays and establishing traceability to higher-order standards. | Critical for demonstrating analytical accuracy and meeting GSPR requirements for metrological traceability, especially for Class C and D devices. |
| Clinical Samples (Retrospective Panels) | Well-characterized, residual patient samples used to determine clinical sensitivity, specificity, and to compare performance against a comparator method. | Must be representative of the target population (age, gender, disease stage, comorbidities). Proper ethical procurement and informed consent documentation are mandatory for the Technical Documentation. |
| Interferent Stocks | Purified substances (e.g., bilirubin, hemoglobin, triglycerides, common drugs) used to spike samples and test for analytical specificity. | Testing a comprehensive panel of potential interferents is required to rule out false results, supporting claims of robustness in the Performance Evaluation Report. |
| Negative/Positive Control Materials | Materials with known negative/positive status used in every assay run to monitor precision and ensure the test is functioning correctly. | Lot-to-lot consistency of control materials must be validated. Their stability defines the assay's reliable operational period and is part of shelf-life studies. |
| Molecular Standards (e.g., gBlocks, RNA transcripts) | Synthetic nucleic acids with known sequences and concentrations used as quantitative standards for PCR-based assays (e.g., for viral load). | Essential for establishing the limit of detection (LoD) and the quantitative range of molecular IVDs. Must be validated against clinical isolates to ensure equivalent detection. |
| Monoclonal/Polyclonal Antibodies (Capture/Detection) | Key components of immunoassays that define the assay's specificity and sensitivity for the target analyte. | Characterized as Analyte Specific Reagents (ASRs). Documentation of source, cloning, purification, and cross-reactivity profiling is critical for the Device Master File. |
| Cell Lines or Viral Lysates | Used as a source of complex native antigens or as a model system for infectious disease assay development. | Helps demonstrate detection of the analyte in a native conformation. Important for validating assays where recombinant proteins alone are insufficient. |
| Stability Study Chambers | Environmental chambers that control temperature and humidity for real-time and accelerated stability testing of reagents and devices. | Data generated is the sole basis for claiming shelf-life on the label, a core IVDR requirement. Protocols must follow recognized standards (e.g., ICH Q1A(R2)). |
The shift from IVDD to IVDR embodies a decisive move towards a more precautionary regulatory model in the EU. This is evidenced by the dramatic expansion of devices requiring Notified Body review, the strict limitation of equivalence routes, and the mandatory, life-cycle based Performance Evaluation. When framed within a US-EU comparative thesis, the IVDR demands a more comprehensive and direct body of evidence prior to market access compared to the common US 510(k) predicate pathway. While the US system offers structured speed via mechanisms like the Breakthrough Devices Program, the EU system prioritizes front-loaded evidence generation. For researchers and developers, this means that strategic planning for the EU market must begin earlier in the development cycle, with a focus on rigorous analytical and clinical study design using well-characterized tools from the scientific toolkit to build a robust Technical Documentation file that satisfies the IVDR's exacting standards.
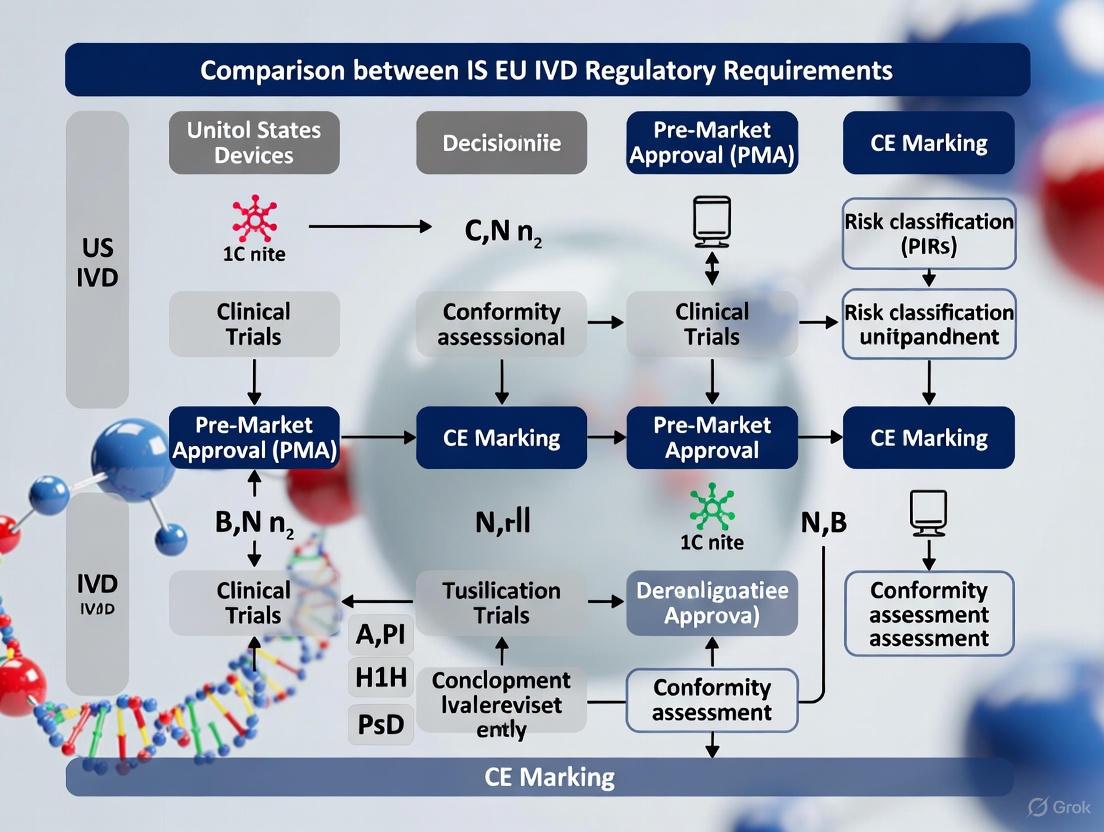
The regulatory pathways for In Vitro Diagnostic (IVD) devices in the United States and the European Union represent two philosophically distinct systems. The US Food and Drug Administration's Center for Devices and Radiological Health (FDA/CDRH) operates as a centralized federal authority with a pro-innovation, risk-based framework. In contrast, the EU employs a decentralized network of Notified Bodies (NBs) under a precautionary regulatory model [6] [1]. This divergence impacts everything from market entry strategy to clinical evidence requirements. Key data illustrates this divide: FDA 510(k) clearance averages 6-12 months, while EU MDR/IVDR certification through a Notified Body typically takes 12-18 months [6]. Furthermore, a 2025 survey indicates that since the implementation of the MDR/IVDR, the choice of the EU as the first launch market has dropped significantly [1]. Understanding these architectures is critical for researchers and developers planning global product commercialization.
Core Regulatory Architectures: Centralized Authority vs. Decentralized Ecosystem
The foundational structures of the US and EU IVD regulatory systems dictate their operational dynamics and strategic implications for manufacturers.
FDA/CDRH (United States): The FDA acts as a singular, federal gatekeeper. Its authority is codified in the Federal Food, Drug, and Cosmetic Act and implemented through the Code of Federal Regulations (21 CFR). The CDRH is responsible for the premarket review, classification, and post-market surveillance of all IVDs marketed in the US. This centralized model aims for consistent application of rules and direct engagement between the agency and industry [6]. The FDA also plays a leading role in global harmonization efforts through its chairmanship of the International Medical Device Regulators Forum (IMDRF), seeking to align regulatory standards internationally [15].
EU Notified Body Ecosystem (European Union): The EU system is decentralized. The European Commission sets the regulations—the In Vitro Diagnostic Regulation (IVDR) 2017/746 and the Medical Device Regulation (MDR) 2017/745—but does not conduct conformity assessments itself. This task is delegated to independent, private organizations designated by individual EU Member States, known as Notified Bodies (NBs) [16]. As of early 2025, there are 51 NBs designated under these regulations [17]. Their capacity is a critical bottleneck; data from mid-2025 shows over 28,489 MDR applications were filed but only 12,177 certificates were issued, with processing times for 60% of cases ranging from 13 to 18 months [1]. A Notified Body’s primary function is to assess a device's conformity with the IVDR's General Safety and Performance Requirements (GSPRs) and issue a CE certificate, which grants market access across the European Economic Area [6] [16].
Table 1: Key Quantitative Metrics of the US and EU IVD Regulatory Systems (Data as of 2025)
| Metric | US (FDA/CDRH) | EU (Notified Body Ecosystem) |
|---|---|---|
| Primary Regulatory Document | 21 CFR (Code of Federal Regulations) | IVDR 2017/746 & MDR 2017/745 |
| Governing Bodies | Single federal agency (FDA) | European Commission + 51 designated Notified Bodies [17] |
| Typical Certification Timeline | 6-12 months (510(k)) [6] | 12-18 months [6] |
| Reported Application-to-Certificate Gap | Not Applicable (Centralized Review) | 28,489 MDR applications vs. 12,177 certificates issued [1] |
| Global Harmonization Role | Chair & Secretariat of IMDRF [15] | Member state participation in IMDRF |
Device Classification Systems
Both systems employ risk-based classification, but the rules and resulting class distributions differ significantly, directly impacting regulatory burden.
FDA Classification: The FDA classifies IVDs as medical devices into three classes (I, II, III) based on intended use and the risk posed to patients. Most IVDs fall into Class II (moderate risk), requiring a 510(k) premarket notification to demonstrate "substantial equivalence" to a legally marketed predicate device. A smaller number of high-risk devices (e.g., companion diagnostics, novel cancer tests) are Class III, requiring a more rigorous Premarket Approval (PMA). Some low-risk Class I devices are exempt from premarket review [6] [3].
IVDR Classification: The IVDR introduces a four-class system (A, B, C, D from low to high risk) based on seven classification rules (Annex VIII). This system dramatically expanded the scope of required Notified Body oversight. Under the old directive, an estimated 80-90% of IVDs were self-certified. Under the IVDR, approximately 80-90% now require a Notified Body review [3]. Class D represents the highest-risk devices (e.g., HIV or Hepatitis B blood screening tests) [18].
Table 2: Comparison of IVD Classification and Associated Pathways
| Risk Level | US FDA Class & Pathway | EU IVDR Class & Pathway | Example IVDs |
|---|---|---|---|
| Lowest Risk | Class I (Mostly Exempt) | Class A (Self-certification) | General culture media, specimen containers |
| Low-Moderate Risk | Class II (510(k)) | Class B (Notified Body Required) | Pregnancy self-tests, cholesterol tests |
| Moderate-High Risk | Class II/III (510(k) or PMA) | Class C (Notified Body Required) | Tumor marker tests, companion diagnostics |
| Highest Risk | Class III (PMA) | Class D (Notified Body Required) | Blood screening for HIV, Hepatitis [18] |
Market Authorization Pathways and Timelines
The procedural journey from application to market authorization highlights the operational contrasts between the two systems.
FDA's 510(k) Pathway (Typical for IVDs): The process is a linear engagement with the FDA. It involves pre-submission meeting (optional but recommended), preparation and submission of the 510(k) dossier, FDA review (with potential Interactive Review cycles), and finally, clearance. The mandated 90-day review clock often extends due to questions, but the path is generally predictable [6].
EU's IVDR Conformity Assessment: The process is negotiated with a chosen Notified Body and involves multiple, often concurrent, steps: device classification, implementation of a quality management system (ISO 13485), preparation of comprehensive technical documentation, Notified Body selection and application, audit (of both QMS and technical documentation), and finally CE certificate issuance. The extended timelines are attributed to NB capacity constraints and, as noted by an EU survey, incomplete manufacturer submissions which contribute to 58% of total processing delays [1]. Furthermore, the IVDR has established a cascade of deadlines for legacy devices, with Class D IVDs requiring a signed agreement with a Notified Body by September 26, 2025, to remain on the market until their full compliance deadline of December 31, 2027 [18].
Diagram 1: Contrasting Centralized vs. Decentralized Regulatory Pathways (Max Width: 760px)
Clinical Evidence and Performance Evaluation Requirements
This area represents one of the most significant divergences in philosophy and requirement.
FDA's Substantial Equivalence: For a 510(k), the core requirement is to demonstrate the new device is as safe and effective as a legally marketed predicate. Clinical data is not always required if analytical and performance testing suffice to prove equivalence. When needed, clinical studies must demonstrate safety and effectiveness for the intended use [6] [3].
IVDR's Performance Evaluation: The IVDR mandates a continuous, iterative process called Performance Evaluation (PE), consisting of three pillars: analytical performance, clinical performance, and scientific validity. A Clinical Performance Study (CPS) is required when existing clinical evidence is insufficient. The requirements for claiming equivalence to an existing device are stricter than the FDA's, demanding similarity not just in intended use but also in technical/biological characteristics and clinical conditions [3]. This often compels manufacturers to generate new clinical data for the EU market.
Table 3: Experimental Protocol Requirements for Clinical Evidence
| Protocol Component | FDA 510(k) Focus | EU IVDR Performance Evaluation Focus |
|---|---|---|
| Primary Objective | Demonstrate substantial equivalence to a predicate device. | Establish analytical & clinical performance per Annex XIII and GSPRs. |
| Study Design | Often focused on comparative testing against the predicate. May require a clinical study for new indications or technologies. | Requires a Performance Evaluation Plan integrating lab data and clinical evidence. Post-Market Performance Follow-up (PMPF) is mandatory for ongoing validation. |
| Data Sources | Predicate device data is central. Literature, in-house lab data, and original clinical data may be used. | Literature, equivalence data (if strict criteria met), and/or original Clinical Performance Studies (CPS). Equivalence is harder to claim. |
| Statistical Analysis | Must support a claim of non-inferiority or equivalence in performance to the predicate. | Must demonstrate safety, scientific validity, and clinical performance throughout the device lifecycle. |
| Post-Market Integration | Part of general post-market surveillance; not formally linked to the pre-submission data plan. | PMPF plan is a required part of the initial technical documentation to proactively collect post-market data [3]. |
Post-Market Surveillance and Vigilance
Both systems enforce robust post-market oversight, but with different structures and reporting mechanisms.
FDA Post-Market Obligations: Manufacturers must comply with Medical Device Reporting (MDR), reporting deaths, serious injuries, and certain malfunctions. They are also subject to Quality System Inspections and must manage changes through submission of new 510(k)s or PMA supplements for significant modifications [6]. A key 2025 update is the transition to the Quality Management System Regulation (QMSR) on February 2, 2026, which harmonizes US QMS requirements with ISO 13485:2016 [19].
IVDR Post-Market Obligations: The IVDR structures requirements by device class. All manufacturers must have a Post-Market Surveillance (PMS) Plan. For Class C and D devices, this results in Periodic Safety Update Reports (PSURs). For Class A and B, a Post-Market Surveillance Report (PMSR) is required [3]. All serious incidents must be reported through the EUDAMED database and may trigger Field Safety Corrective Actions (FSCAs). The system emphasizes proactive trend reporting and continuous update of the Performance Evaluation Report [6].
The Emerging-Tech Divide: AI/ML and Cybersecurity
The regulatory approach to software and digital health technologies is rapidly diverging, creating a new strategic frontier.
AI/ML-Enabled Devices: The US has adopted a pro-innovation stance. The FDA's Predetermined Change Control Plan (PCCP) guidance allows developers to pre-specify and gain approval for future AI model modifications (like re-training) within a controlled plan, avoiding the need for a new submission with each iteration [1] [19]. This aligns with a broader national AI strategy aimed at reducing regulatory burdens [1]. In the EU, AI-enabled IVDs face a dual regulatory burden: they must comply with both the IVDR and the EU AI Act. Most medical AI systems are classified as high-risk, requiring conformity assessment under both frameworks, which demands rigorous risk management, data governance, and transparency documentation [1].
Cybersecurity: The FDA's updated 2025 guidance mandates a Secure Product Development Framework (SPDF) be built into the product lifecycle, requiring a Software Bill of Materials (SBOM) and vulnerability management plans in premarket submissions [1]. In the EU, cybersecurity requirements are spread across multiple laws: the IVDR's GSPRs, the NIS 2 Directive, the AI Act, and the Radio Equipment Directive (RED), creating a complex, multi-layered compliance challenge [1].
Strategic Analysis and Future Outlook
The regulatory divide necessitates informed market-entry strategies.
Strategic Pathways: A "US-First" strategy is increasingly common, particularly for innovative or AI-driven devices, due to faster potential access, clearer digital health pathways, and the ability to generate revenue and real-world evidence to support a subsequent EU application [1]. An "EU-First" strategy may be preferable for devices with strong existing clinical data, when targeting global markets that recognize CE marking, or for companies with established EU manufacturing [6].
Future Outlook: The EU system is under review for potential simplification. The European Commission launched a "Call for Evidence" in late 2025 to gather stakeholder input on reducing administrative burdens and improving predictability, indicating awareness of the system's challenges [9]. In the US, the focus is on enhancing predictability and global alignment through the QMSR transition and ongoing IMDRF leadership [19] [15].
Table 4: Strategic Analysis for IVD Market Entry Planning
| Decision Factor | Favor US-First Strategy | Favor EU-First Strategy |
|---|---|---|
| Primary Target Market | Revenue primarily from the US market [6]. | Revenue targeted across EU and global markets recognizing CE mark [6]. |
| Technology Profile | AI/ML-enabled devices (PCCP pathway) [1]; Devices with a clear US predicate. | Devices with robust existing clinical data meeting IVDR equivalence rules. |
| Regulatory Resources | Limited initial budget for comprehensive clinical studies. | Capability to invest in extensive clinical performance studies and technical documentation. |
| Time-to-Market Urgency | Faster initial commercial launch is critical [6]. | Longer timeline is acceptable for broader market access foundation. |
| Manufacturing Base | Manufacturing located in or supplying the US. | Manufacturing located within the EU/EEA. |
The Scientist's Toolkit: Essential Reagents & Materials for IVD Development & Validation
This table outlines critical materials used in the experiments and evaluations required for regulatory submissions.
Table 5: Key Research Reagent Solutions for IVD Development
| Item | Primary Function in IVD Development/Validation |
|---|---|
| Recombinant Antigens & Antibodies | Serve as critical raw materials for assay development; used as calibrators, controls, and capture/detection molecules. Their purity and specificity are central to analytical performance studies. |
| Clinical Specimen Panels | Well-characterized human samples (serum, plasma, tissue) used to establish clinical sensitivity, specificity, and accuracy during clinical performance studies. |
| Reference Standard Materials | Traceable standards (e.g., WHO International Standards) used to validate assay accuracy, precision, and establish the clinical reportable range. |
| Linearity & Dilution Panels | Materials used to demonstrate assay performance across its measuring range, a key component of analytical validation for quantitative tests. |
| Interference Substances | Substances (e.g., bilirubin, lipids, common drugs) used to test and document assay specificity and potential interfering substances. |
| Stability Testing Materials | Reagents and specimens used to conduct real-time and accelerated stability studies to support claimed shelf-life and in-use stability. |
Diagram 2: IVDR Performance Evaluation & Clinical Evidence Lifecycle (Max Width: 760px)
This comparison guide analyzes the risk-based classification systems for In Vitro Diagnostic (IVD) medical devices in the United States (US) and European Union (EU). Framed within broader research on US versus EU IVD regulatory requirements, this guide objectively compares the structures, application rules, and compliance consequences of the US Food and Drug Administration's (FDA) three-class system and the EU's In Vitro Diagnostic Regulation (IVDR) four-class system [20] [21]. Understanding these frameworks is critical for researchers, scientists, and drug development professionals navigating global market access, as the classification dictates the rigor of premarket review, clinical evidence requirements, and ongoing post-market surveillance [22] [23].
Core Comparison of IVD Risk Classification Systems
The US and EU systems are both founded on the principle of proportional risk but differ in structure, specific rules, and the resulting distribution of devices across classes.
Table: Comparison of US FDA and EU IVDR Risk Classification Systems
| Aspect | US FDA System | EU IVDR System |
|---|---|---|
| Governing Regulation | 21 CFR Parts 800-898 [23] | Regulation (EU) 2017/746 (IVDR) [20] [22] |
| Risk Classes | Class I (Low), Class II (Moderate), Class III (High) [24]. | Class A (Low), Class B (Moderate), Class C (High), Class D (Highest) [20] [21]. |
| Basis for Classification | Risk to patient/individual based on intended use and indications for use. Primarily determined by finding a "substantially equivalent" predicate device [24] [21]. | Risk to patient/individual and public health [20]. Governed by 7 classification rules in IVDR Annex VIII based on device type and intended purpose [20] [21]. |
| Typical Examples | Class I: General lab instruments [21]. Class II: Pregnancy tests, cholesterol tests [24]. Class III: HIV blood donor screening tests, high-risk companion diagnostics. | Class A: Specimen containers, buffers [20]. Class B: Pregnancy, fertility, cholesterol self-tests [20] [21]. Class C: HIV/STD tests, genetic tests, companion diagnostics [20] [21] [25]. Class D: Blood/ tissue donor screening for life-threatening transmissible agents (e.g., HIV, HCV) [20] [21]. |
| Key Classification Rules | Predicate-based comparison. Class III defined as life-supporting/sustaining, of substantial importance in preventing impairment, or presenting potential unreasonable risk [24]. | Rule 1: High-risk transmissible agents -> Class D [20]. Rule 2: Blood/tissue compatibility -> Class C [20]. Rule 3: Infectious agents, cancer testing -> Class C [20]. Rule 4: Self-testing -> Class C (exceptions like pregnancy are Class B) [20]. Rule 5: General lab products -> Class A [20]. Rule 6: Default class -> Class B [20]. |
| Primary Regulatory Pathway | Class I: Most exempt from premarket notification [510(k)] [24]. Class II: Requires 510(k) clearance (demonstration of substantial equivalence) [24]. Class III: Requires Premarket Approval (PMA) [24]. | Class A (non-sterile): Self-declaration by manufacturer [22] [25]. Class A (sterile), B, C, D: Requires conformity assessment by a Notified Body [20] [22]. Class D requires the most stringent assessment [25]. |
| % of IVDs Requiring 3rd Party Review (Notified Body/FDA) | Majority of Class I and all Class II/III undergo FDA review [24]. | Estimated 80-90% of IVDs require Notified Body review under IVDR [20]. |
Regulatory Pathways and Conformity Assessment
The risk classification directly determines the conformity assessment route a device must take to obtain market authorization (FDA clearance/approval or CE Marking).
Table: Conformity Assessment Pathways by Device Class
| Device Class | US FDA Pathway | EU IVDR Pathway (for CE Marking) |
|---|---|---|
| Low Risk (US I / EU A) | Most are exempt from premarket submission [24]. Must still register establishment and list device. Subject to general controls (e.g., labeling, GMP). | Class A non-sterile: Manufacturer self-declares conformity [22] [25]. No Notified Body audit required. Class A sterile: Notified Body audits sterility aspects only [22]. |
| Moderate Risk (US II / EU B) | Premarket Notification [510(k)]: Requires demonstration of substantial equivalence to a predicate device [24]. | Full Notified Body Assessment: Audit of Quality Management System (QMS) and technical documentation review [22] [25]. |
| High Risk (US III / EU C) | Premarket Approval (PMA): Requires submission of scientific evidence (often clinical trial data) to demonstrate safety and effectiveness [24]. | Full Notified Body Assessment: More stringent scrutiny of technical documentation and clinical evidence [25]. For Companion Diagnostics (Class C), Notified Body must consult with a medicines agency (e.g., EMA) [25]. |
| Highest Risk (EU D only) | N/A (US Class III is the highest). | Full Notified Body Assessment: Most stringent level of scrutiny. Specific rules for devices detecting life-threatening transmissible agents [20] [25]. |
Experimental Data and Performance Evaluation Requirements
Both jurisdictions require robust clinical or performance evidence, scaled to the device's risk class. The IVDR formalizes this as a "performance evaluation" consisting of three pillars [22].
Methodology 1: Conducting an IVDR Performance Evaluation The Performance Evaluation Report (PER) is mandatory for CE Marking under IVDR [22].
- Objective: To demonstrate scientific validity, analytical performance, and clinical performance of an IVD [22].
- Protocol:
- Scientific Validity: Establish that the analyte (e.g., biomarker) is associated with the clinical condition. Evidence can come from literature, peer-reviewed publications, or clinical trial data [22].
- Analytical Performance: Verify performance metrics like accuracy, precision, sensitivity, specificity, and limits of detection. Studies are conducted using contrived or clinical samples [23].
- Clinical Performance: Establish the device's ability to yield results correlated to a clinical condition. For novel high-risk devices (Class C/D), this typically requires a performance study (clinical investigation) [26]. For lower-risk or well-established devices, existing literature or results from equivalent devices may be used [27].
- Data Analysis: Statistical plans must be pre-specified. For clinical performance, metrics like Positive/Negative Percent Agreement (PPA/NPA) with a reference method or clinical endpoint are standard [26].
Methodology 2: Leveraging Real-World Data (RWD) for Performance Studies RWD is increasingly used to support regulatory submissions, especially for post-market studies or when traditional clinical trials are impractical [23].
- Objective: To generate clinical evidence on device performance from routine healthcare data.
- Protocol:
- Study Design: Define a retrospective or prospective cohort using electronic health records, disease registries, or biobank samples [23].
- Sample Selection: Apply clear inclusion/exclusion criteria. A key challenge is managing competition for scarce clinical samples, especially for rare diseases [23].
- Data Collection & Validation: Ensure the IVD results from leftover or prospectively collected samples are linked to validated clinical endpoints. Data provenance and quality control are critical [26] [23].
- Statistical Analysis: Account for potential biases (selection, information) in the RWD. Methods like propensity score matching may be used to strengthen the evidence [23].
Logical Workflow Diagrams
US IVD Classification Decision Workflow
EU IVD Classification Rule Application Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table: Key Materials for IVD Development and Validation
| Item | Function in IVD Development/Evaluation | Key Regulatory Consideration |
|---|---|---|
| Well-Characterized Clinical Samples | Used to establish clinical sensitivity/specificity during performance evaluation. Includes positive, negative, and borderline specimens [23]. | Scarcity of rare disease samples is a major challenge. Provenance, informed consent, and stability data are critical for regulatory acceptance [26] [23]. |
| Reference Materials & Standards | Provide a benchmark for calibrating assays and verifying analytical performance (precision, accuracy) [20]. | Use of internationally recognized standards (e.g., WHO) strengthens regulatory submissions. For novel biomarkers, creating an in-house primary reference may be necessary. |
| Contrived Samples | Artificially created samples spiked with a target analyte at known concentrations. Used for analytical validation when clinical samples are scarce [23]. | Must be scientifically justified. The matrix should closely mimic the native clinical sample to demonstrate comparable assay performance [23]. |
| Quality Management System (QMS) Software | Manages the entire device lifecycle documentation required for regulatory submissions (e.g., design controls, risk management, technical files) [20] [22]. | Under IVDR, a compliant QMS is mandatory for all classes [22] [25]. Software like Greenlight Guru or SimplerQMS helps structure documentation for audit readiness [20] [22]. |
| Unique Device Identifier (UDI) System | A standardized system for device identification and traceability throughout its distribution and use [22] [25]. | Mandatory for CE Marking (to be registered in EUDAMED) and for US FDA registration. Facilitates post-market surveillance and vigilance activities [22] [23]. |
A significant strategic shift is underway in the global in vitro diagnostics (IVD) industry, with a growing number of manufacturers prioritizing the United States market over the European Union for initial product launches. This trend is a direct consequence of a deepening regulatory divide. The U.S. Food and Drug Administration (FDA) maintains a more predictable and pro-innovation framework, while the EU's In Vitro Diagnostic Regulation (IVDR) presents a complex, resource-intensive, and capacity-constrained pathway [1]. Quantitative analysis reveals a striking market impact: since the implementation of the IVDR, the selection of the EU as the first launch market has decreased by approximately 40% for large IVD manufacturers [1]. This comparison guide objectively analyzes the regulatory data and strategic implications behind this pivotal trend, providing researchers and development professionals with the evidence needed to inform global launch strategies.
Comparative Analysis of US and EU IVD Regulatory Frameworks
The decision for a US-first launch strategy is rooted in fundamental differences between the regulatory ecosystems. The following tables break down these critical distinctions.
Table 1: Foundational Regulatory Structure Comparison
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Regulated as medical devices under 21 CFR; LDTs currently under CLIA oversight [3] [23]. | Regulated under standalone In Vitro Diagnostic Regulation (EU) 2017/746 [3] [11]. |
| Risk Classification | 3 classes: I (lowest risk), II, III (highest risk). Dictates regulatory pathway [3]. | 4 classes: A (lowest risk), B, C, D (highest risk). Defined by Annex VIII rules [3] [28]. |
| Notified Body Involvement | Not applicable. FDA conducts reviews. | Required for approximately 80% of IVDs (Classes B, C, D, and sterile A), a major increase from ~20% under the old directive [1] [28]. |
| Core Review Standard | Substantial Equivalence: Demonstration of equivalence to a legally marketed predicate device is often sufficient [1]. | Exact Equivalence: Stricter requirements for demonstrating equivalence to an existing device, creating a higher barrier [1]. |
| AI/ML-Enabled Device Pathway | Predetermined Change Control Plans (PCCP): Allows pre-approved, iterative updates to AI models without a new submission each time, supporting agile development [1] [23]. | Dual Regulation: AI-based IVDs must comply with both IVDR and the EU AI Act, typically as "high-risk" systems, requiring integrated conformity assessment [1]. |
Table 2: Market Pathway and Evidence Requirements
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Key Premarket Pathways | 510(k) (Class I/II), De Novo (novel, low-moderate risk), PMA (Class III) [3]. | Conformity Assessment via Notified Body (Annexes IX-XI), leading to CE marking [3]. |
| Clinical Evidence Focus | Required for Class III and some Class II devices; focused on safety and effectiveness for intended use [3]. | Performance Evaluation (PE): Mandatory for all devices, based on three pillars: Scientific Validity, Analytical Performance, and Clinical Performance [3]. |
| Post-Market Surveillance (PMS) | Mandatory reporting of adverse events and malfunctions [3]. | Structured, risk-proportionate system: Periodic Safety Update Reports (PSUR) for Class C/D, Post-Market Surveillance Reports (PMSR) for Class A/B [3]. |
| Cybersecurity Requirements | Lifecycle approach via Secure Product Development Framework (SPDF). Requires Software Bill of Materials (SBOM) in premarket submissions [1] [23]. | Multi-layered requirements under IVDR General Safety and Performance Requirements (GSPRs), NIS2 Directive, AI Act, and Radio Equipment Directive [1]. |
| Unique Device Identification (UDI) | Required. Managed via FDA's GUDID system [3]. | Required. Format differs from US; data stored in the European Database on Medical Devices (EUDAMED) [3] [23]. |
Experimental Protocols for Regulatory Evidence Generation
Successful market authorization in any region requires robust experimental data. The following protocols outline standard methodologies for generating the analytical and clinical evidence central to regulatory dossiers.
Protocol 1: Analytical Performance Study for a Quantitative IVD This protocol is designed to fulfill core analytical performance requirements under both FDA and IVDR frameworks.
- Objective: To determine the precision, accuracy, linearity, and limit of detection (LoD) of a novel immunoassay for Serum Biomarker X.
- Materials:
- Candidate assay reagents and calibrators.
- Control materials at low, medium, and high concentrations.
- Clinical serum samples (retained, patient-deidentified).
- Predicate or comparator assay (if claiming equivalence).
- Automated clinical chemistry analyzer.
- Experimental Design:
- Precision: Perform 20 replicates of low, medium, and high controls over 5 days (total 100 data points per level) to calculate within-run, between-run, and total coefficient of variation (%CV) [3].
- Accuracy/Method Comparison: Analyze 120 clinical samples with a concentration spread across the assay's measuring interval using both the candidate and predicate method. Perform Passing-Bablok regression and Bland-Altman analysis.
- Linearity: Prepare a series of dilutions from a high-concentration sample. Analyze replicates to confirm the measured value is directly proportional to the dilution factor across the claimed range.
- Limit of Detection (LoD): Analyze a minimum of 20 replicates of a zero calibrator (or sample with no analyte). Calculate LoD as mean + 2SD (or per CLSI EP17 guideline).
- Data Analysis: All statistical analyses (mean, SD, %CV, regression) must be performed using validated software (e.g., SAS, R). Acceptance criteria (e.g., total CV <10%, correlation coefficient R² >0.95) must be pre-defined based on intended use and state of the art.
Protocol 2: Clinical Performance Study for a High-Risk (Class C/D or PMA) IVD This protocol generates the clinical validity and performance data essential for IVDR Performance Evaluation and FDA PMA applications.
- Objective: To evaluate the clinical sensitivity and specificity of a novel molecular diagnostic test for Genetic Mutation Y in colorectal cancer patients.
- Study Design: Prospective, multi-center, observational study.
- Subject Population:
- Cases: 200 patients with confirmed colorectal cancer (CRC).
- Controls: 200 individuals undergoing colonoscopy with no evidence of neoplasia (healthy controls) or with benign polyps.
- Inclusion/Exclusion criteria must be meticulously defined.
- Sample Collection & Testing:
- Collect matched formalin-fixed, paraffin-embedded (FFPE) tumor tissue and whole blood from each participant.
- Test all samples using the index test (the novel assay) under investigation.
- Test all samples using the reference standard. For genetic tests, this is typically validated Next-Generation Sequencing (NGS) performed at a CAP/CLIA-certified laboratory [23].
- Blinding: Index test operators must be blinded to the reference standard results and clinical diagnosis, and vice versa.
- Primary Endpoints:
- Clinical Sensitivity: (True Positives / (True Positives + False Negatives)) x 100.
- Clinical Specificity: (True Negatives / (True Negatives + False Positives)) x 100.
- 95% confidence intervals must be calculated for both metrics.
- Statistical Plan: A pre-specified statistical analysis plan must include sample size justification based on power analysis to detect a minimum performance threshold.
Visualizing Regulatory Pathways and Strategic Decisions
Diagram 1: Comparative US FDA vs. EU IVDR Approval Pathways
Diagram 2: Decision Logic for US-First Launch Strategy
Table 3: Key Research and Compliance Resources
| Tool / Resource | Function / Purpose | Application in IVD Development |
|---|---|---|
| Clinical Samples (Retained, Biobanked) | Provide the biological matrix for analytical and clinical performance studies [23]. | Essential for method comparison (Protocol 1) and establishing clinical sensitivity/specificity (Protocol 2). Scarcity is a major challenge for rare diseases [23]. |
| Predicate/Comparator Device | Serves as a benchmark for establishing substantial equivalence (US) or for comparative performance evaluation (EU/US) [1]. | Central to the 510(k) pathway and often used as the reference method in analytical protocols. |
| Validated Reference Standard | Provides the "ground truth" for clinical performance studies, often a gold-standard method or clinically confirmed diagnosis. | Critical for Protocol 2. For genetic tests, this is often orthogonal NGS or Sanger sequencing performed in an accredited lab. |
| ISO 13485:2016 Quality System | Provides the framework for a Quality Management System (QMS) harmonized with regulatory requirements [28]. | Mandatory for EU conformity assessment and aligned with FDA's Quality System Regulation (QMSR). Governs all design, development, and production controls. |
| EUDAMED (European Database) | The EU's centralized database for device registration, UDI, Notified Body certificates, clinical investigations, and post-market surveillance [3] [23]. | Required for CE marking. Manufacturers must register and upload device data; modules become mandatory from 2026 [23]. |
| NIST AI Risk Management Framework (RMF) | A voluntary framework for managing risks in AI systems, focusing on governance, mapping, measuring, and managing risk [1]. | Provides a flexible, best-practice structure for developing trustworthy AI/ML components in IVDs, supporting FDA submissions. |
| Software Bill of Materials (SBOM) | A formal, machine-readable inventory of software components, libraries, and dependencies [1] [23]. | A cybersecurity requirement for "cyber device" submissions to the FDA and a best practice for EU IVDR compliance to manage vulnerability risks. |
| Common Specifications (CS) | EU documents providing detailed technical/clinical requirements for specific high-risk device types (e.g., Class D IVDs) [11]. | Define the state of the art and mandatory performance criteria for devices like HIV or HBV blood screening tests, guiding protocol design. |
Strategic Implications and Future Outlook
The data-driven comparison confirms that the US-first trend is a rational industry response to divergent regulatory pressures. The US offers a faster, more predictable route to initial revenue, which is crucial for funding further development and evidence generation. The ability to use a PCCP for AI/ML devices is a particular accelerator for next-generation diagnostics [1]. Conversely, the EU IVDR, while aiming for high safety, acts as a significant brake on speed to market due to Notified Body bottlenecks, stringent evidence requirements, and layered regulations for AI and cybersecurity [1] [28].
However, Europe remains a critical market with an estimated value of USD 34.47 billion by 2033 [29]. Therefore, the dominant strategy is not to abandon Europe, but to sequence market entry: launch first in the U.S., gather real-world evidence and revenue, and then leverage that data to support the more demanding EU submission [1]. This approach mitigates risk and optimizes resource allocation.
The landscape is dynamic. The EU Commission has initiated a "Call for Evidence" to explore simplifying regulations [1], while the UK is developing its own framework that may recognize approvals from peer regulators like the FDA [23]. Professionals must monitor these developments closely, as any future harmonization or mutual recognition could reshape the strategic calculus for global IVD launches.
Practical Pathways: Navigating Premarket Approval and Compliance
In vitro diagnostic (IVD) devices in the United States are regulated by the Food and Drug Administration (FDA) as medical devices and are subject to a risk-based classification system that determines the premarket review pathway [3]. The three primary pathways are the Premarket Notification (510(k)), the De Novo Classification, and the Premarket Approval (PMA). These pathways exist within a global regulatory landscape, most notably contrasted with the European Union's In Vitro Diagnostic Regulation (IVDR) [3]. Understanding these pathways—their strategic applications, evidence requirements, and outcomes—is critical for researchers and developers aiming to efficiently bring innovative diagnostic tools to the U.S. market while planning for global commercialization.
Comparative Analysis of US Premarket Pathways
The choice between the 510(k), De Novo, and PMA pathways is fundamentally dictated by the novelty of the device and its risk classification, which the FDA defines as Class I (low to moderate risk), Class II (moderate to high risk), or Class III (high risk) [30] [31].
The volume of submissions, associated costs, and review timelines for each pathway vary significantly, reflecting their differing levels of regulatory scrutiny.
Table 1: Pathway Utilization and Financial Costs (FY 2024-2025) [30] [31]
| Regulatory Pathway | Risk Class | Applications Received (FY2024) | Standard FDA User Fee (FY2025) | Small Business User Fee (FY2025) | FDA Review Clock Goal |
|---|---|---|---|---|---|
| 510(k) | Class I/II (some III) | 3,643 [30] | $24,335 [31] | $6,084 [31] | ~90 days [31] |
| De Novo | Class I/II | 78 [30] | $162,235 [30] | $40,559 [30] | 150 days [30] |
| PMA | Class III | 69 [30] | $540,783 [30] | $135,196 [30] | 180+ days |
Substantive Requirements and Evidence Thresholds
The scientific and technical evidence required for a successful submission differs markedly between the pathways, scaling with the perceived risk and novelty of the device.
Table 2: Key Regulatory and Evidence Requirements [30] [4] [32]
| Requirement | 510(k) Pathway | De Novo Pathway | PMA Pathway |
|---|---|---|---|
| Core Regulatory Basis | Demonstration of Substantial Equivalence (SE) to a predicate device [4]. | Risk-based classification of a novel device with no predicate [30] [32]. | Demonstration of safety and effectiveness for high-risk devices [30]. |
| Predicate Device | Required. Must be a legally marketed U.S. device [4]. | Not applicable; pathway is for devices without a predicate [31]. | Not applicable; intended for novel high-risk devices. |
| Clinical Data | Not routinely required. May be needed if technological differences raise new safety questions [31]. | Often required; approximately 80% of submissions include clinical studies [30]. | Extensively required. Comprehensive clinical investigation data is the cornerstone of the application [30]. |
| Benefit-Risk Analysis | Implicit in SE determination. | Explicitly required to justify classification and controls [31]. | Extensive, rigorous assessment required. |
| Post-Market Change Flexibility | Moderate. Follows 510(k) change guidance; many changes require a new submission [30]. | High initially, then follows 510(k) rules. Once granted, the device becomes a predicate for 510(k)s, and future modifications follow 510(k) standards [30]. | Very low. Almost all changes require prior FDA approval via a PMA supplement [30]. |
| Outcome | Clearance via an SE Letter [31]. | Authorization via a Grant Order, creating a new classification and product code [31]. | Approval via a PMA Grant Order [30]. |
Pathway Decision Logic
Selecting the appropriate pathway is a critical strategic decision. The following diagram outlines the key decision points based on device novelty and risk.
Detailed Examination of Individual Pathways
The 510(k) Premarket Notification Pathway
The 510(k) is the most common regulatory pathway, accounting for thousands of clearances annually [30]. It is suitable for devices that can demonstrate substantial equivalence (SE) to a predicate device legally marketed in the U.S. prior to May 28, 1976 (a "preamendments device"), or a device cleared through 510(k) or De Novo since [4].
- Substantial Equivalence Definition: A device is SE if it has the same intended use and the same technological characteristics as the predicate. If the technological characteristics differ, the submitter must show the differences do not raise new questions of safety and effectiveness and that the device is as safe and effective as the predicate [4].
- Evidence Standard: The focus is on comparative testing against the predicate. While clinical data is not routinely required, comprehensive bench performance data (e.g., analytical sensitivity, specificity, precision, stability) is essential [31].
- Outcome and Impact: A cleared 510(k) does not create a new regulatory category. The device is classified according to its predicate. Post-market changes are subject to evaluation, with significant changes requiring a new 510(k) [30].
The De Novo Classification Pathway
The De Novo pathway, created to foster innovation, is for novel devices of low to moderate risk that lack a predicate [32]. It prevents such devices from being automatically relegated to the high-burden PMA pathway.
- Strategic Value: A successful De Novo request results in an authorization to market and, more importantly, creates a new regulatory classification with a unique product code and special controls [31]. This device then serves as a predicate for future 510(k) submissions, offering a "first-mover" advantage [30] [32].
- Evidence Standard: Unlike 510(k), the sponsor must provide valid scientific evidence (often including clinical data) to establish a reasonable assurance of safety and effectiveness for the novel device type [30] [31]. A benefit-risk analysis is mandatory.
- Access Routes: There are two ways to access the De Novo pathway: 1) After a Not Substantially Equivalent (NSE) determination for a 510(k), or 2) via a Direct De Novo request when the sponsor determines from the outset that no predicate exists [31].
The Premarket Approval (PMA) Pathway
PMA is the most stringent pathway, reserved for Class III high-risk devices [30]. These are devices that support or sustain human life, are of substantial importance in preventing health impairment, or present a potential unreasonable risk of illness or injury.
- Evidence Standard: PMA applications require the most comprehensive evidence, including extensive clinical investigation data from well-controlled studies [30]. The application must provide a full accounting of the device's design, manufacturing, labeling, and a complete benefit-risk profile.
- Review Process: The review is intensive and interactive, often involving an advisory panel of external experts. All aspects of manufacturing are scrutinized, and a pre-approval inspection of manufacturing facilities is typical [30].
- Post-Market Control: Post-approval requirements are heavy, including mandatory post-approval studies and stringent control over device changes, nearly all of which require prior FDA approval via a PMA supplement [30].
Experimental Protocols for Clinical Evidence Generation
The generation of robust clinical evidence is central to the De Novo and PMA pathways and is increasingly relevant for certain 510(k)s. The methodology must be tailored to the device's intended use and claims.
Protocol Design and Regulatory Considerations
Objective: To generate clinical performance data (e.g., sensitivity, specificity, positive/negative predictive value) sufficient to support the regulatory claim of safety and effectiveness.
Core Methodology:
- Study Design: Prospective, retrospective, or cross-sectional study designs are employed using clinically relevant samples. The patient population must be representative of the intended-use population [33].
- Comparator Method: Performance is compared to a gold standard reference method (for efficacy) and/or to existing standard-of-care diagnostic procedures (for usability and clinical impact).
- Endpoint Definition: Primary endpoints (e.g., clinical sensitivity/specificity) and secondary endpoints (e.g., reproducibility, usability) are pre-specified and aligned with the device's stated claims.
- Statistical Analysis Plan: A pre-defined plan details sample size calculation (powered to demonstrate performance within a pre-specified confidence interval), analysis populations, and methods for handling missing data.
Regulatory Oversight (U.S.):
- Studies intended to support a PMA are considered significant risk investigations and require an Investigational Device Exemption (IDE) approved by an Institutional Review Board (IRB) and the FDA before initiation [4].
- Many De Novo and some 510(k) studies may also require an IDE. Sponsors are strongly advised to engage with the FDA via the Q-Submission (Q-Sub) process to align on study design and data requirements before beginning [31].
EU-IVDR Comparison: Performance Studies
The EU's IVDR demands a structured Performance Evaluation for all IVDs, consisting of three pillars: scientific validity, analytical performance, and clinical performance [3]. The process for generating clinical evidence is governed by specific articles of the IVDR.
Key Divergences from U.S. Process:
- Oversight Body: Clinical evidence is reviewed by a Notified Body (third-party auditor) rather than directly by the central authority [3] [33]. For companion diagnostics, an additional consultation with the European Medicines Agency (EMA) is mandatory [33].
- Study Notification/Application: The IVDR categorizes performance studies into tiers requiring either a notification to or an application approved by the relevant national Competent Authorities and Ethics Committees, based on the study's invasiveness and risk [34] [26].
- Standard: The state-of-the-art standard for conducting studies is ISO 20916 (Good Study Practice for IVDs), which differs from the principles commonly referenced in U.S. submissions [34].
The Scientist's Toolkit: Essential Reagents & Materials
Successfully navigating premarket submissions requires meticulous preparation of technical documentation and performance data.
Table 3: Key Research Reagent Solutions and Regulatory Materials
| Item / Category | Function in IVD Development & Submission | Relevant Pathway(s) |
|---|---|---|
| Well-Characterized Clinical Samples | Biobanked or prospectively collected specimens with confirmed disease status (via reference method). Used for clinical validation studies to establish sensitivity, specificity, and predictive values. | Primary: De Novo, PMA. Critical for 510(k) if claiming superiority or new indications. |
| Reference Standard Materials | Highly purified analytes, international standard panels, or standardized control materials. Used to establish analytical sensitivity (LoD), calibration, and cross-reactivity panels. | All Pathways. Foundational for analytical performance testing. |
| Predicate Device(s) & Data | For 510(k), the legally marketed device(s) for which substantial equivalence is claimed. Access to its labeling and publicly available performance data is essential for comparison. | Primary: 510(k). |
| ISO 20916:2024 Framework | The European standard specifying Good Study Practice for planning, conducting, and reporting IVD performance studies. Essential for structuring clinical investigations for the EU market and increasingly recognized as a best practice globally [34]. | Primary: IVDR (EU). Informative for U.S. studies. |
| Q-Submission (Q-Sub) Package | A structured request for feedback from the FDA on proposed study designs, data requirements, or regulatory pathway. Not a reagent, but a critical procedural tool to de-risk development [31]. | All U.S. Pathways, especially De Novo and PMA. |
| Performance Evaluation Report (PER) | The comprehensive document required under IVDR that integrates data on scientific validity, analytical performance, and clinical performance of the device [3]. | Primary: IVDR (EU). Analogous to the clinical evidence summary in U.S. submissions. |
Analysis within US vs. EU Regulatory Thesis
A comparative analysis of U.S. and EU IVD regulatory requirements reveals a complex landscape of convergence in scientific expectations and divergence in process and oversight.
Table 4: Core Thesis Comparison: US FDA Pathways vs. EU IVDR [3] [33]
| Aspect | United States (FDA) | European Union (IVDR) | Implications for Global Strategy |
|---|---|---|---|
| Regulatory Philosophy | Centralized pre-market review by the FDA, with depth scaling by risk class (510(k), De Novo, PMA). | Conformity assessment based on meeting General Safety & Performance Requirements (GSPRs), often via a Notified Body. Pre-market scrutiny scales with class (A-D). | U.S. has a single gatekeeper; EU involves third-party audits, adding a layer of complexity in partner management. |
| Risk Classification | 3 Classes (I, II, III). IVDs are medical devices. Class dictates pathway [3]. | 4 Classes (A, B, C, D). Specific rules in Annex VIII of IVDR. Companion Diagnostics are Class C [3] [33]. | Direct alignment is impossible. A device may be Class II (510(k)) in the U.S. but Class C (Notified Body review) in the EU. |
| Evidence of Clinical Validity | Required for PMA and most De Novos. For 510(k), triggered by new questions of safety/effectiveness [31]. | Required for all classes via the Performance Evaluation, with depth proportional to risk [3]. | The EU mandates a formalized Performance Evaluation process for all devices, while the U.S. process is more pathway-dependent. |
| Companion Diagnostic Pathway | Evolving. Recent proposals seek to reclassify many NGS/NAAT oncology CDx from Class III (PMA) to Class II (510(k) with special controls), potentially streamlining review [33]. | Fixed as Class C. Requires Notified Body assessment + mandatory consultation with EMA (or national authority), creating a multi-actor, potentially high-friction pathway [33]. | A significant divergence. The U.S. may become a faster, less costly market for certain mature CDx technologies, altering global launch sequencing. |
| Post-Market Surveillance | Medical Device Reporting (MDR), registries for some devices, post-approval studies for PMAs. | Systematic, structured, and proportional to risk. Periodic Safety Update Reports (PSURs) for Class C/D devices and Post-Market Performance Follow-up (PMPF) plans are required [3]. | EU requirements are more prescriptive and uniformly applied across high-risk devices compared to the U.S. |
Conclusion for Researchers: While both regions demand strong analytical and clinical performance evidence, the regulatory mechanics differ profoundly. The U.S. system is pathway-driven with a central regulator, whereas the EU employs a risk-rule-driven, distributed model with mandatory third-party assessment. For novel IVDs, the U.S. De Novo and EU IVDR Class C/D routes both demand rigorous clinical evidence, but the operational workload, timelines, and costs associated with Notified Body reviews and EMA consultations can make the EU pathway particularly demanding [33]. Successful global development requires building a unified evidence package that can be adapted to meet the specific procedural requirements of each jurisdiction.
The European Union's In Vitro Diagnostic Regulation (IVDR) 2017/746 represents a fundamental and stringent overhaul of the regulatory landscape for diagnostic devices [35]. For researchers and developers accustomed to the former In Vitro Diagnostic Directive (IVDD) or the U.S. Food and Drug Administration (FDA) system, engaging with Notified Bodies (NBs)—the independent third-party organizations designated to assess conformity—is now a critical and complex step for market access [16] [36]. This guide compares the IVDR's conformity assessment paradigm with the U.S. FDA framework, providing a data-driven analysis for professionals navigating both jurisdictions.
The IVDR Paradigm Shift: From Self-Certification to Notified Body Scrutiny
Under the previous IVDD, an estimated 80-90% of IVDs were self-certified by manufacturers, requiring no independent review [35]. The IVDR has inverted this model. Now, approximately 80-90% of IVDs require a conformity assessment by a Notified Body [35] [37]. This shift exponentially increases the number of devices and manufacturers needing NB engagement, creating significant capacity challenges [17].
A 2025 survey of all 51 EU-designated Notified Bodies highlights the ongoing tension between application volumes and certification output [17]. While applications for both the Medical Device Regulation (MDR) and IVDR continue to rise, a persistent gap between applications submitted and certificates issued indicates systemic capacity constraints [17]. For IVDs specifically, upward trends are noted for Class B and C devices, which fall under mandatory NB assessment [17]. Delays are attributed to complex review processes and incomplete technical documentation from manufacturers [17].
Table 1: Impact of the IVDR Transition on Notified Body Involvement
| Regulatory Framework | % of IVDs Requiring Notified Body Assessment | Key Consequence for Manufacturers |
|---|---|---|
| EU IVDD (Previous) | 10-20% [35] | Majority of devices could be self-declared; limited NB interaction. |
| EU IVDR (Current) | 80-90% [35] [37] | Vast majority require formal NB audit and certification; strategic NB partnership is essential. |
| U.S. FDA | 100% (via FDA review) | Centralized authority; all market submissions require FDA review (e.g., 510(k), PMA) [10]. |
Comparative Analysis: EU IVDR vs. U.S. FDA Regulatory Pathways
A core element of the broader US vs. EU regulatory thesis lies in their foundational structures: the EU employs a decentralized network of independent Notified Bodies, while the U.S. operates a centralized governmental authority (the FDA) [14].
Regulatory Oversight and Classification
The pathways to market and the entities governing them differ substantially.
Table 2: Comparison of Regulatory Oversight and Classification
| Aspect | U.S. FDA IVD Regulation | EU IVDR |
|---|---|---|
| Oversight Body | Centralized (FDA/Center for Devices and Radiological Health) [14] [10]. | Decentralized (Notified Bodies designated by EU Member States) [16] [14]. |
| Primary Premarket Pathways | Premarket Notification [510(k)] (demonstration of substantial equivalence); De Novo (novel, low-to-moderate risk); Premarket Approval (PMA) (high risk) [10]. | Conformity Assessment via Notified Body (Annexes IX-XI of IVDR). Routes involve QMS audit and technical documentation review [36] [38]. |
| Classification System | Class I (low), II (moderate), III (high) [10]. Risk-based, with classifications codified in 21 CFR [10]. | Class A (lowest), B, C, D (highest) [37]. Rule-based (7 rules in Annex VIII), considering patient and public health risk [35] [37]. |
| Basis for Decision | FDA review team makes regulatory decision based on submitted data [10]. | Notified Body performs assessment and issues certificate; authority rests with the NB [16] [36]. |
Experimental Protocol: Determining the Correct Classification and Pathway
- Intended Use Analysis: Precisely define the device's intended use, target population, and diagnostic claim.
- Rule Application (EU): Apply the seven classification rules in IVDR Annex VIII sequentially [37]. For example, a device detecting a sexually transmitted agent (Rule 3) or a blood-borne pathogen (Rule 1) would be classified as Class C or D, respectively [37].
- Predicate Identification (US): Research existing, legally marketed devices for potential substantial equivalence. If no predicate exists, the De Novo or PMA pathway must be considered [10].
- Regulatory Strategy Documentation: Justify the determined classification and premarket pathway in a regulatory strategy document, referencing the specific rules (EU) or regulation citations (US).
Requirements for Clinical and Performance Evidence
Both regions demand robust evidence but differ in structure, terminology, and ongoing requirements.
Table 3: Comparison of Evidence Requirements
| Aspect | U.S. FDA IVD Regulation | EU IVDR |
|---|---|---|
| Core Requirement | Safety and effectiveness for the intended use [10]. | Demonstration of safety, performance, and scientific validity [35]. |
| Premarket Focus | Analytical and clinical validation data included in 510(k), De Novo, or PMA submission [10]. Emphasis on pre-market study data. | Performance Evaluation Report (PER), comprising scientific validity, analytical performance, and clinical performance data [35]. |
| Post-Market Focus | General post-market surveillance; reporting of adverse events and malfunctions [14] [35]. | Continuous requirement for post-market performance follow-up (PMPF). Mandatory Periodic Safety Update Report (PSUR) for Class C & D devices and Post-Market Surveillance Report (PMSR) for Class A & B [35]. |
| Equivalence Pathway | 510(k) is based on demonstration of substantial equivalence to a predicate device [10]. | Use of equivalent device data is possible but severely restricted under IVDR. Requires full access to underlying data and demonstration of same technical, biological, and clinical characteristics [14] [35]. |
Experimental Protocol: Building a Performance Evaluation Report (PER) for IVDR
- Scientific Validity: Establish the association between the analyte (marker) and the clinical condition. Protocol: Conduct a systematic literature review to gather and appraise existing scientific evidence linking the analyte to the disease/condition [37].
- Analytical Performance: Verify the device's measurable performance characteristics. Protocol: Design method validation studies to assess accuracy, precision, sensitivity, specificity, detection limit, measuring range, and robustness per CLSI/ISO standards.
- Clinical Performance: Demonstrate the device's ability to yield results correlating with a clinical condition. Protocol: Execute a clinical performance study (or analyze existing data) to determine clinical sensitivity and specificity. For high-risk (Class D) devices, this often requires a prospective clinical investigation.
- Report Synthesis: Analyze all data in a benefit-risk context and compile into the PER, which is a core part of the technical documentation reviewed by the Notified Body [35].
Post-Market Surveillance and Vigilance
The IVDR mandates a more proactive and structured post-market surveillance (PMS) system than the FDA's typically more reactive system [35].
The Engagement Process with a Notified Body
Engaging with an NB under IVDR is a multi-stage, iterative process distinct from a single submission to the FDA.
Diagram: The IVDR Conformity Assessment & CE Marking Process [36] [38]
Critical Consideration: Notified Body Capacity and Selection The 2025 survey underscores that NB capacity is a bottleneck [17]. Researchers must:
- Initiate early: Contact NBs 12-24 months before the desired certification date.
- Prepare thoroughly: Incomplete documentation is a major cause of delay [17]. A comprehensive, IVDR-ready technical file is essential.
- Choose strategically: Consider an NB's specific designations, expertise in your device type, and geographic location.
Successfully navigating the IVDR requires leveraging specific tools and resources.
Table 4: Key Research Reagent Solutions & Regulatory Tools
| Tool / Resource | Function / Purpose | Relevance to IVDR Compliance |
|---|---|---|
| EUDAMED (European Database on Medical Devices) | Central EU database for actor registration, UDI, certificates, vigilance, and clinical investigations [35]. | Mandatory for registering economic operators, uploading Summary of Safety and Performance (SSP), and vigilance reporting. |
| Harmonized Standards (e.g., ISO 14971:2019, ISO 20916) | Published standards whose reference is in the Official Journal of the EU, providing presumption of conformity to IVDR requirements [35]. | State-of-the-art application of ISO 14971 for risk management and ISO 20916 for clinical performance studies is expected by NBs [35]. |
| NANDO Database | Official EU database listing all designated Notified Bodies and their scope of designation [16]. | Critical for verifying an NB's legal designation to assess your specific device class and type. |
| Performance Evaluation Software (e.g., DistillerSR) | Software for managing systematic literature reviews for scientific validity and clinical evaluation [37]. | Essential for producing the reproducible, auditable literature reviews required for the Performance Evaluation Report. |
| IVDR Classification Rule Guidance | Guidance documents interpreting the seven classification rules in Annex VIII [37]. | Foundational for determining the correct risk class (A-D), which dictates the conformity assessment pathway. |
Foundational Regulatory Frameworks and Classification
The technical documentation for an in vitro diagnostic (IVD) is a comprehensive record that demonstrates a product’s safety and performance. Its structure and evidentiary requirements are dictated by the foundational regulations of the target market. In the United States, the Food and Drug Administration (FDA) regulates IVDs as medical devices under the Federal Food, Drug, and Cosmetic Act, primarily guided by 21 CFR Parts 807, 809, and 820 [10] [39]. In the European Union, IVDs are governed by a dedicated regulation, the In Vitro Diagnostic Regulation (IVDR, EU 2017/746), which repealed the previous directive [11].
Both regions employ a risk-based classification system, but the structures differ, directly impacting the depth of the technical file and the rigor of its review.
- US FDA Classification: IVDs are classified into Class I (lowest risk), II, or III (highest risk) [3] [10]. The classification determines the premarket submission pathway: most Class I devices are exempt from review, Class II typically requires a 510(k) demonstrating substantial equivalence, and Class III requires Premarket Approval (PMA) [10] [39].
- EU IVDR Classification: IVDs are classified under four risk-based rules (Annex VIII) into Class A (lowest risk), B, C, or D (highest risk) [3]. A critical change from the previous directive is that an estimated 80-90% of IVDs now require the involvement of a Notified Body for certification, compared to about 10-20% before [3] [39]. Class D devices face the most stringent oversight, including potential review by an EU reference laboratory [11].
The following diagram illustrates how the classification dictates the regulatory and documentation pathway in each jurisdiction.
Figure 1: Risk classification and its impact on the regulatory pathway for IVDs in the US and EU.
Technical Documentation: Structure and Core Requirements
The compilation of evidence is known by different terms but serves the same fundamental purpose. In the EU IVDR, it is explicitly called "technical documentation" (Annexes II and III) [40]. In the US, under the Quality System Regulation (QSR), the primary compilation was historically the Device Master Record (DMR), though the FDA's alignment with ISO 13485 through the new Quality Management System Regulation (QMSR) is moving terminology toward the "medical device file" [40] [39]. Both systems require a detailed design history (Design History File or design and development files) [40].
The core elements required in the technical file show significant alignment but with key philosophical differences, as summarized in the table below.
Table 1: Core Elements of IVD Technical Documentation: US vs. EU Requirements
| Document Element | United States (FDA) | European Union (IVDR) | Key Comparative Note |
|---|---|---|---|
| Device Description & Intended Use | Required in DMR/Device File [40]. | Required (Annex II, Ch. I) [40]. | Closely aligned. |
| Essential Principles / General Safety & Performance Requirements (GSPRs) | General controls and special controls (for Class II/III) [10]. | Must demonstrate conformity with Annex I GSPRs [3]. | IVDR structure is more explicit and enumerated. |
| Risk Management File | Required per 21 CFR 820.30 & .100 [39]. | Required per ISO 14971 [39]. | Both reference the same standard (ISO 14971). |
| Design & Manufacturing Information | DMR contains specifications and procedures [40]. | Required (Annex II, Ch. II & III) [40]. | Highly similar in intent. |
| Verification & Validation Evidence | Required for design controls (21 CFR 820.30) [40]. | Required, including analytical and clinical performance data [3] [40]. | IVDR requires a structured Performance Evaluation Report (PER) [41] [39]. |
| Clinical Evidence | Required for PMA; for 510(k), often analytical studies with clinical samples suffice [10]. | Mandatory Performance Evaluation (Annex XIII) for all classes, including scientific validity, analytical & clinical performance [3] [39]. | EU requires continuous, lifecycle-based evidence generation for all IVDs. |
| Post-Market Surveillance Plan | Implied in complaint handling and MDR reporting (21 CFR 803) [39]. | Explicitly required (Annex III). Must produce Periodic Safety Update Reports (PSURs) for Class C&D or Post-Market Surveillance Reports (PMSRs) for Class A&B [3] [39]. | EU has a more formalized and periodic reporting structure. |
| Labeling & Instructions for Use | Must comply with 21 CFR 809.10 (IVD-specific) [3] [10]. | Must comply with IVDR Annex I, Chapter IV. | Both are prescriptive; labels must be region-specific [39]. |
| Declaration of Conformity | Not applicable. | Required to affix CE marking [40]. | Unique to the EU system. |
A critical divergence is in the lifecycle management of the file. The FDA's review is generally tied to premarket submission milestones, while the IVDR embeds continuous updating of the technical documentation and the Performance Evaluation as part of post-market surveillance [3] [39].
Generating Evidence: Experimental Protocols and Performance Evaluation
The experimental data populating the technical file must be generated under rigorous methodologies. The required evidence differs in scope between the regions, particularly for clinical validity.
1. Analytical Performance Studies (US & EU): These are foundational for both regions and demonstrate an IVD's technical capability.
- Objective: To determine the test's analytical sensitivity (detection limit), analytical specificity (interference), accuracy, precision, and measuring range [10].
- Protocol Summary: Testing is performed using well-characterized samples (e.g., clinical samples, reference standards, or contrived samples). Experiments follow CLSI guideline methodologies. Precision is assessed over multiple days, operators, and instrument lots. Analytical sensitivity is often determined via a dilution series of a known positive sample.
- Data Presentation: Results are summarized with descriptive statistics (mean, standard deviation, coefficient of variation) and presented via plots (e.g., linearity plots, precision profiles, ROC curves for limit of detection studies).
2. US 510(k) Substantial Equivalence Studies: For a 510(k) submission, the goal is to demonstrate the new device is as safe and effective as a legally marketed predicate [10].
- Objective: To show comparable analytical performance to a predicate device.
- Protocol Summary: A method comparison study is conducted by testing a set of clinical samples (typically 100+), representative of the intended use population and covering the assay's measuring range, on both the new and predicate devices [10].
- Data Presentation: Data is analyzed via correlation (e.g., Pearson's r) and difference plots (Bland-Altman). Statistical tests for bias (e.g., paired t-test) are performed. Clinical sensitivity/specificity may be calculated if applicable [10].
3. EU IVDR Performance Evaluation (Annex XIII): This is a comprehensive, three-pillar process unique to the IVDR that must be documented in the PER [3].
- Pillar 1: Scientific Validity: Evidence that the analyte (marker) is associated with the clinical condition.
- Protocol: A literature review and/or analysis of existing clinical data from biobanks or databases.
- Pillar 2: Analytical Performance (as described above).
- Pillar 3: Clinical Performance: Evidence the device correctly identifies or predicts the clinical condition in the target population.
- Protocol: A clinical performance study, which may be a prospective trial, a retrospective study using archived samples, or a literature review. For novel markers, a prospective study is typically required. Samples must be representative of the EU population [39].
- Data Presentation: Calculation of clinical sensitivity, specificity, positive/negative predictive values, likelihood ratios, and their confidence intervals. For quantitative tests, measures of agreement (e.g., hazard ratios in outcome studies) are presented.
Table 2: Comparison of Key Experimental Evidence Requirements
| Evidence Type | US FDA (for 510(k)) | EU IVDR | Primary Objective |
|---|---|---|---|
| Analytical Performance | Required [10]. | Required (Pillar 2) [3]. | Establish technical reliability. |
| Clinical Comparison Study | Often required using clinical samples vs. a predicate [10]. | Not applicable in the US form. | Demonstrate equivalence to predicate. |
| Scientific Validity | Not a formal requirement. | Required (Pillar 1) [3]. | Establish link between marker and disease. |
| Clinical Performance Study | Not routinely required for 510(k); expected for PMA [10]. | Required (Pillar 3) for all IVDs (scale proportional to risk) [3] [39]. | Establish diagnostic accuracy in target population. |
| Post-Market Performance Follow-up (PMPF) | Ad hoc via complaint investigation. | Required to update the Performance Evaluation [3]. | Continuously confirm safety and performance. |
The Scientist's Toolkit: Essential Reagents & Materials for IVD Performance Studies
- Well-Characterized Clinical Samples: Residual patient samples or samples from biobanks, with associated laboratory and clinical data. Function: The gold-standard material for analytical and clinical performance studies [10].
- International Standard Reference Materials: Certified reference materials from bodies like WHO or NIBSC. Function: To establish traceability, calibrate assays, and demonstrate accuracy [11].
- Commercial Quality Control Materials: Assayed controls at multiple levels. Function: To monitor precision and stability during validation studies and routine testing.
- Interferent & Cross-Reactivity Panels: Solutions of common endogenous (bilirubin, hemoglobin, lipids) and exogenous (medications) substances, and related analytes. Function: To rigorously assess analytical specificity [10].
- DNA/RNA Panels (for molecular tests): Characterized panels with sequences covering genetic variants, polymorphisms, and near-neighbor strains. Function: To validate assay inclusivity (detection of all targets) and exclusivity (no cross-reaction).
Strategic Implementation for Global Market Access
For researchers targeting both markets, strategic planning is essential.
- Adopt a Global QMS Framework: Implement a quality management system based on ISO 13485:2016, which is mandatory for the EU and increasingly aligned with FDA requirements under the QMSR [39]. Integrate ISO 14971 for risk management.
- Design Studies for Dual Compliance: Plan analytical performance studies to meet the highest standard required by either region. For clinical evidence, design studies that can satisfy the FDA's predicate comparison needs and generate the clinical performance data (sensitivity, specificity) required for the IVDR's PER [10] [39]. Using the IMDRF Table of Contents as a backbone for the technical file can help structure documentation for multiple regulators [40].
- Engage with Regulators Early: Use the FDA's Pre-Submission (Q-Sub) process to get feedback on novel test designs or complex study protocols [10]. In the EU, early dialogue with a Notified Body is crucial, especially for higher-class devices.
- Plan for Lifecycle Management: From the start, establish processes for the continuous update of clinical evidence and technical documentation as required by the IVDR, integrating these activities into the post-market surveillance system [3] [39].
The global in vitro diagnostics (IVD) market, projected to reach USD 197.06 billion by 2032, operates under two distinct regulatory paradigms: the United States (US) Food and Drug Administration (FDA) framework and the European Union (EU) In Vitro Diagnostic Regulation (IVDR) [42]. For researchers and developers, navigating the divergent demands for clinical evidence and performance evaluation is critical for market access. The US system, where IVDs are regulated as medical devices, employs a risk-based classification (I, II, III) that dictates evidence requirements [3]. In contrast, the EU's IVDR (Regulation (EU) 2017/746) establishes a self-contained, four-class risk system (A, B, C, D) and mandates a continuous, three-pillar Performance Evaluation for all devices [3] [43]. This guide provides a detailed, data-driven comparison of these requirements, offering clear protocols and insights for strategic evidence generation.
Comparative Analysis of Regulatory Pathways and Evidence Demands
Foundational Regulatory Structures
- United States (FDA): IVDs are classified as medical devices under the Code of Federal Regulations (CFR). The pathway—510(k) clearance, De Novo request, or Premarket Approval (PMA)—is determined by the device's risk class (I, II, or III) and the existence of a valid predicate [3].
- European Union (IVDR): IVDs are regulated under a dedicated framework separate from general medical devices. Market access requires a CE marking following a conformity assessment, the rigor of which is dictated by the device's risk class (A, B, C, D). A notified body is involved for most classes (B, C, D and sterile Class A) [3] [44].
Side-by-Side Comparison of Key Elements
Table 1: Comparison of US and EU IVD Regulatory Fundamentals
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Food, Drug & Cosmetic Act; 21 CFR Parts 800-898 [3] | Regulation (EU) 2017/746 [11] |
| Risk Classification | 3 Classes (I, II, III) [3] | 4 Classes (A, B, C, D) [3] |
| Market Authority | Food and Drug Administration (FDA) | Notified Bodies (for most classes) & Competent Authorities [3] |
| Core Evidence Concept | Clinical evidence required based on classification and submission type [3]. | Performance Evaluation (PE): A continuous process based on three pillars [43]. |
| Evidence Requirements | Varies: Often focused on analytical and clinical performance for the intended use. | Mandatory for all classes: 1) Scientific Validity, 2) Analytical Performance, 3) Clinical Performance [43]. |
| Post-Market Surveillance | Adverse event reporting (e.g., MAUDE). Periodic reports for PMA devices [3]. | Structured system: PMS Plan, PMPF Plan, Periodic Safety Update Reports (PSURs for C&D), Post-Market Surveillance Reports [3] [43]. |
| Unique Device Identification (UDI) | Required. Managed via the FDA's GUDID database [3]. | Required. Format differs from US; data stored in EUDAMED [3]. |
| Transition/"Grandfathering" | Predicate-based 510(k) allows reliance on prior devices [3]. | No grandfathering. Staggered transition deadlines (2025-2027) from old Directive to IVDR [44]. |
Quantitative Market Context
Table 2: IVD Market Snapshot and Growth Projections
| Region | Projected 2025 Market Revenue | Projected CAGR (2025-2032/2030) | Key Driver |
|---|---|---|---|
| Global | USD 126.73 Bn [42] | 6.5% (to 2032) [42] | Rising chronic/infectious disease burden; personalized medicine [42]. |
| North America | USD 35.28 Bn [45] | 3.22% (to 2030) [45] | High healthcare expenditure; advanced tech adoption; strong payer systems [45]. |
| United States | USD 32.15 Bn [45] | (Part of NA CAGR) | Dominant global player (44.3% 2025 share) [42]. |
| European Union | (Part of Global) | (Part of Global) | Implementation of IVDR shaping market dynamics and evidence standards [44]. |
Core Experimental Protocols for Performance Evaluation
The EU IVDR's structured Performance Evaluation (PE) provides a comprehensive model for generating robust evidence [43]. The following protocols detail the methodologies for its three pillars.
Protocol 1: Establishing Scientific Validity
- Objective: To demonstrate the well-established association between the analyte (biomarker) and the clinical condition or physiological state.
- Methodology – Systematic Literature Review (SLR):
- Protocol Development: Define the review question (PICO format), inclusion/exclusion criteria, and search strategy.
- Literature Search: Execute searches in multiple databases (e.g., PubMed, Embase, Cochrane Library). Document the search strategy and results.
- Study Selection & Data Extraction: Screen results against criteria. Extract data on the biomarker's biological role, pathophysiological mechanism, and clinical association from relevant studies.
- Evidence Synthesis & Reporting: Critically appraise the quality of evidence. Synthesize findings to build a scientifically valid argument. Compile a Scientific Validity Report (SVR) justifying the analyte's link to the intended use [43].
Protocol 2: Assessing Analytical Performance
- Objective: To verify the device's ability to accurately, precisely, and reliably detect or measure the analyte under specified conditions [43].
- Methodology – Controlled Laboratory Studies: Experiments must be conducted using protocols aligned with IVDR Annex II, Section 6.1 and relevant standards (e.g., CLSI EP guidelines) [43].
- Key Experiments & Acceptance Criteria:
- Accuracy/Trueness: Comparison of method results to a reference method or certified reference material. Calculate bias and recovery.
- Precision: Repeatability (within-run) and reproducibility (between-day, operator, instrument) studies. Report as standard deviation (SD) and coefficient of variation (%CV).
- Analytical Sensitivity (Limit of Detection, LoD): Determine the lowest analyte concentration reliably distinguished from blank. Use low-concentration samples and a defined statistical method (e.g., 2SD or 95% confidence).
- Analytical Specificity: Test for interference (e.g., hemoglobin, lipids, bilirubin) and cross-reactivity with structurally similar substances.
- Measuring Range & Linearity: Test samples across the claimed range. Assess linearity through polynomial regression.
- Deliverable: An Analytical Performance Report (APR) summarizing all validation data against pre-defined specifications [43].
Protocol 3: Demonstrating Clinical Performance
- Objective: To confirm the device provides clinically actionable and reliable results in the target population within the intended use setting.
- Methodology – Clinical Performance Study:
- Study Design: Prospective, retrospective, or cross-sectional study design appropriate to the claim. Define primary endpoints (e.g., clinical sensitivity/specificity, Positive/Negative Predictive Value).
- Subject Selection: Enroll subjects from the intended population. Include clear inclusion/exclusion criteria.
- Sample Testing & Comparator: Test subject samples with the device under evaluation. Compare results to a clinical reference standard (e.g., diagnostic gold standard, well-established comparator assay, clinical consensus).
- Statistical Analysis: Calculate performance metrics with confidence intervals (e.g., 95% CI). Use statistical tests (e.g., ROC analysis) to establish or verify cut-offs [43].
- Deliverable: A Clinical Performance Report (CPR) documenting the study and proving diagnostic accuracy and clinical utility [43].
The Scientist's Toolkit: Essential Research Reagent Solutions
Generating regulatory-grade evidence requires high-quality, traceable materials. This toolkit lists essential solutions for executing the protocols above.
Table 3: Essential Research Reagent Solutions for IVD Performance Evaluation
| Reagent/Material | Primary Function in Evaluation | Critical Quality Attribute |
|---|---|---|
| Certified Reference Materials (CRMs) | Serves as the primary standard for establishing accuracy (trueness) and metrological traceability [43]. | Certified value with stated uncertainty, traceability to SI units or international standard. |
| Biobanked Clinical Specimens | Provides real-world matrix for analytical and clinical studies. Used for precision, interference, and clinical sensitivity/specificity testing [26]. | Well-characterized disease state/analyte concentration, ethical sourcing, stability data. |
| Interference & Cross-Reactivity Panels | Challenges the assay to prove analytical specificity. Contains high concentrations of potential interferents and structurally similar analytes [43]. | Purity, relevance to intended use setting (e.g., common medications, disease-state molecules). |
| Calibrators and Quality Controls | Used in daily runs to establish and monitor the assay's measuring scale and stability. Critical for robustness and longitudinal performance data [43]. | Value assignment traceable to CRM, low commutability bias, long-term stability. |
| DNA/RNA Reference Panels | For molecular assays, these provide characterized sequences for establishing analytical sensitivity (LoD) and specificity, including for genetic variants [43]. | Sequence-verified, quantified, and matched to claimed genomic targets. |
Visualizing Regulatory Pathways and Evidence Generation
The following diagrams map the logical flow of regulatory decision-making and evidence integration under both systems.
Diagram 1: US FDA vs EU IVDR Regulatory Decision Pathways (Max Width: 760px)
Diagram 2: The EU IVDR Performance Evaluation Lifecycle (Max Width: 760px)
Strategic Implications for Researchers and Developers
The comparison reveals strategic imperatives for global product development:
- Evidence Generation Planning: An IVDR-compliant Performance Evaluation Plan (PEP) is the most comprehensive starting point, as it addresses scientific, analytical, and clinical pillars. Evidence generated for the EU can often be adapted for FDA submissions, but the reverse is not always true, especially regarding scientific validity justification [43].
- Study Design for Dual Submissions: When planning studies, incorporate EU reference standards and population demographics early to ensure data acceptability in both regions. The EU's requirement for a Post-Market Performance Follow-up (PMPF) plan means clinical data collection must be designed as a continuous activity, not a one-time pre-market effort [3] [43].
- Navigating Recent Developments: The June 2025 release of MDCG 2025-5, a Q&A guidance on performance studies, provides critical clarification for study planning in the EU [26] [46]. Furthermore, the European Commission has initiated a "Call for Evidence" for a targeted revision of the IVDR, signaling an ongoing evolution of the framework aimed at reducing administrative burden while maintaining safety [9]. Researchers must monitor these updates, as they may impact future evidence strategies.
The global market for In Vitro Diagnostic (IVD) devices operates under two predominant but historically divergent regulatory philosophies: the United States' Food and Drug Administration (FDA) framework and the European Union's In Vitro Diagnostic Regulation (IVDR 2017/746). Central to both are mandated Quality Management Systems (QMS)—the FDA's Quality System Regulation (QSR, 21 CFR Part 820) and the internationally recognized ISO 13485:2016 standard [10] [47]. For researchers and developers, understanding their comparison is critical for strategic planning and global market access.
A significant harmonization shift is underway. The FDA has issued a final rule to amend its QSR, renaming it the Quality Management System Regulation (QMSR) and incorporating by reference the requirements of ISO 13485:2016 [47]. This rule becomes effective and enforceable on February 2, 2026 [47]. Concurrently, within the EU, ISO 13485 is a harmonized standard under the IVDR, providing a presumption of conformity with the regulation's QMS requirements, although full IVDR compliance demands additional specific processes [48] [49]. This analysis compares these systems within a thesis on US vs. EU IVD requirements, focusing on structural nuances, experimental evidence requirements, and implications for development professionals navigating this converging landscape.
Comparative Analysis of Core QMS Requirements
The foundational requirements for design, risk management, and production controls share common goals but differ in specific emphasis and structure between the current FDA QSR and ISO 13485.
Design Controls: Both frameworks mandate rigorous design and development processes. The FDA QSR provides a highly prescriptive, subsystem-oriented approach with detailed requirements for design inputs, outputs, review, verification, and validation [50]. ISO 13485 also requires design controls but structures them within a process-oriented model, integrating them with planning, review, and change control [51]. Recent FDA enforcement has focused on "510(k) drift"—where marketed devices differ from cleared designs—and tracing post-market complaints back to inadequate design inputs, indicating intense scrutiny in this area [50].
Risk Management: ISO 13485 integrates risk-based thinking throughout the QMS, requiring organizations to apply a risk-based approach to control processes. It is typically implemented alongside ISO 14971, the dedicated risk management standard [48]. The FDA QSR implicitly requires risk management primarily within design controls (820.30) and corrective actions (820.100) [10]. The IVDR explicitly emphasizes risk management and a favorable benefit-risk ratio, requiring alignment with ISO 14971 within the QMS [52].
Production and Process Controls: Both systems enforce controls for production, inspection, and traceability. A key enforcement trend in 2025 is inadequate oversight of contract manufacturers (CMOs). The FDA holds sponsors accountable for CMO actions, citing issues like poor segregation and lack of oversight in shared facilities [50]. ISO 13485 explicitly addresses control of outsourced processes, requiring defined criteria and controls [49].
Table 1: Comparison of Key QMS Requirement Emphases
| QMS Area | FDA QSR (21 CFR 820) | ISO 13485:2016 | IVDR Context |
|---|---|---|---|
| Overall Structure | Subsystem-based (Design, CAPA, etc.) [50] | Process-oriented, integrated system [51] | Requires QMS per Article 10(9); EN ISO 13485 is harmonized [48] [49] |
| Risk Management | Focused within Design Controls & CAPA [10] | Risk-based thinking throughout QMS; aligned with ISO 14971 [52] | Explicit requirement for benefit-risk; must integrate ISO 14971 [52] |
| Design & Development | Highly prescriptive, detailed stages [50] | Required, but within process model [51] | Technical documentation (Annex II/III) must demonstrate performance [52] |
| Supplier Control | Purchasing Controls (820.50) [50] | Control of outsourced processes & suppliers [49] | Critical supplier control is emphasized [52] |
| Management Responsibility | Management with executive responsibility [10] | Management commitment, resource provision [53] | Includes Person Responsible for Regulatory Compliance (PRRC) [52] |
| Post-Market Surveillance | Complaint Handling (820.198), MDR [50] | Feedback, complaint handling, reporting [49] | Comprehensive PMS plan and Post-Market Performance Follow-up (PMPF) required [49] |
Harmonization Trajectory: FDA QMSR and Global Alignment
The regulatory landscape is moving toward harmonization, with the FDA's adoption of ISO 13485 as the cornerstone.
The FDA QMSR Final Rule: Published in February 2024, this rule amends 21 CFR Part 820 to incorporate ISO 13485:2016 by reference, with an effective date of February 2, 2026 [47]. The FDA states the requirements are "substantially similar" but adds clarifying expectations to ensure consistency with other FDA statutes [47]. This transition represents a significant cultural and procedural shift for both industry and regulators, moving from a checklist-based QSIT inspection model to a process-based audit approach [51].
Global Recognition of ISO 13485: The standard is a global regulatory foundation. It is integral to the Medical Device Single Audit Program (MDSAP) and is a designated standard in the UK [53]. Its position is under review, with a systematic review by ISO scheduled for 2025 to decide on confirmation or revision [53].
Table 2: Key Dates in Quality System Harmonization
| Date | Regulatory Event | Implication for IVD Manufacturers |
|---|---|---|
| February 2, 2024 | FDA publishes QMSR Final Rule [47] | Official start of 2-year transition from QSR to QMSR. |
| May 26, 2025 | IVDR Deadline for QMS for Class D legacy devices [49] | Legacy Class D IVDs must have an IVDR-compliant QMS to lodge application. |
| 2025 | ISO Systematic Review of ISO 13485:2016 [53] | Decision point on whether to confirm or revise the standard. |
| February 2, 2026 | FDA QMSR becomes enforceable [47] | All FDA inspections will assess compliance with QMSR (ISO 13485). |
Experimental Protocols and Validation Requirements
Demonstrating compliance requires robust experimental data. Both US and EU pathways demand rigorous validation, though the emphasis and context differ.
Analytical and Clinical Performance Validation: For FDA 510(k) clearance, reviews focus on analytical performance characteristics (bias, imprecision, specificity, sensitivity) compared to a predicate [10]. Clinical samples are typically used. For higher-risk devices, clinical performance data demonstrating sensitivity and specificity is required [10]. Under the IVDR, the performance evaluation is a continuous process requiring scientific validity, and analytical and clinical performance data, forming the core of clinical evidence [49].
Emergency Use Pathways: The FDA has finalized guidance for emergency use of IVDs during public health crises under Section 564 of the FD&C Act [54]. It emphasizes a benefit-risk assessment, the urgency of need, and risk mitigation for false results. This requires pre-validated data packages and robust QMS controls for rapid deployment [54].
Example Protocol: Analytical Validation for a Quantitative IVD Assay
This protocol outlines key experiments to establish performance claims for regulatory submissions.
1. Objective: To determine the precision, accuracy, linearity, reportable range, and specificity of [Assay Name] for the quantification of [Analyte] in [Sample Matrix].
2. Experimental Design:
- Precision: Perform within-run (20 replicates of 3 samples spanning low, mid, high concentrations) and between-run (duplicate tests of 3 samples over 20 days) experiments per CLSI EP05-A3.
- Accuracy/Method Comparison: Test ≥100 patient samples spanning the measuring range against a validated comparator method (e.g., FDA-cleared assay). Perform correlation and bias analysis (Passing-Bablok regression, Bland-Altman plots).
- Linearity & Reportable Range: Prepare a series of ≥5 spiked samples covering the claimed range. Analyze each sample in triplicate. Assess linearity via polynomial regression.
- Analytical Specificity: Test for interference from common substances (e.g., bilirubin, hemoglobin, lipids) per CLSI EP07. Assess cross-reactivity with structurally similar analytes.
3. Data Analysis & Acceptance Criteria: Predefined criteria (e.g., total imprecision <15%, bias <10%, correlation R² >0.98) must be based on intended clinical use and state-of-the-art. Out-of-specification results trigger investigation per QMS CAPA procedures [50].
The Scientist's Toolkit for IVD Development and Compliance
Table 3: Essential Research Reagent Solutions and Regulatory Tools
| Tool/Reagent Category | Specific Example/Function | Role in IVD Development & Compliance |
|---|---|---|
| Reference Standards & Controls | WHO International Standards, NIST SRMs, Certified Reference Materials (CRMs) | Establish traceability, calibrate assays, validate accuracy. Critical for generating credible analytical performance data [10]. |
| Clinical Sample Panels | Characterized, disease-state serum/plasma panels; remnant patient samples. | Used for clinical validation studies to establish sensitivity, specificity, and clinical accuracy [10] [49]. |
| Interferent Stocks | Purified bilirubin, intralipid solutions, hemoglobin lysates. | For conducting interference testing to demonstrate analytical specificity as part of validation protocols. |
| Documentation & e-Submission Tools | FDA eSTAR (Electronic Submission Template) [55] | Interactive PDF required for 510(k) and De Novo submissions. Guides structured data entry, automates checks, and improves submission quality [55]. |
| Risk Management Software | Tools aligned with ISO 14971 framework. | Facilitates hazard analysis, risk estimation, and control tracking. Essential for IVDR compliance and integrated QMS risk management [52]. |
| Performance Evaluation Templates | PEP (Performance Evaluation Plan), PER (Report), SCP (Summary of Clinical Performance) templates. | Structured documents required under IVDR to plan, execute, and report on the performance evaluation for clinical evidence [49]. |
Visualization of Regulatory Harmonization and QMS Transition
Diagram 1 Title: Convergence of US and EU IVD quality system pathways toward an ISO 13485 baseline.
Diagram 2 Title: QMS transition workflow for FDA QMSR compliance by February 2026.
The comparison between the FDA's QSR and ISO 13485 is rapidly evolving from one of divergence to strategic convergence. The FDA's adoption of ISO 13485 via the QMSR significantly aligns the US with global regulatory practices, reducing duplicative requirements for manufacturers seeking market access in multiple regions [53] [47].
For IVD researchers and developers, this means:
- Building a unified QMS foundation based on ISO 13485 is the most efficient strategy for global compliance, requiring overlay of specific regional requirements (e.g., FDA's added clauses, IVDR's performance evaluation and PRRC roles) [47] [52].
- Preparation for a cultural shift in regulatory interactions is essential, particularly for the FDA's move from a subsystem checklist (QSIT) to a process-based audit model [50] [51].
- Robust, data-driven design and validation remain paramount. While the QMS framework harmonizes, the experimental burden for proving safety, efficacy, and performance is not diminished. Recent FDA trends show intense scrutiny on CAPA effectiveness, design control rigor, and complaint handling [50]. Under the IVDR, the continuous generation of clinical evidence through performance evaluation is a central and demanding requirement [49].
- Proactive transition is critical. Manufacturers must begin their QMS transition now to meet the FDA's February 2026 deadline and the IVDR's phased QMS deadlines for legacy devices, the first being May 2025 for Class D [47] [49].
The overarching thesis of US-EU IVD regulatory comparison thus finds a unique nexus in quality systems: despite different regulatory philosophies on pre-market review, post-market surveillance rigor, and classification rules, the foundational system for ensuring ongoing quality is unifying around a common international standard, signaling a significant step toward regulatory harmonization.
Overcoming Implementation Hurdles and Optimizing Regulatory Strategy
This comparison guide examines the operational and strategic challenges posed by the European Union's In Vitro Diagnostic Regulation (IVDR), with a specific focus on the bottleneck created by limited Notified Body (NB) capacity and complex transition deadlines. Framed within a broader thesis contrasting U.S. and EU regulatory philosophies, this analysis provides researchers and drug development professionals with data-driven insights to navigate the divergent pathways to market for IVDs. The U.S. Food and Drug Administration (FDA) is characterized by a pro-innovation, risk-based approach that often serves as a catalyst for early market entry [1] [56]. In stark contrast, the EU's IVDR embodies a precautionary principle, creating a more complex environment where conformity assessment capacity has become a critical constraint on market access [1]. Understanding this divide is essential for global strategic planning.
Comparative Analysis of Notified Body Capacity and Certification Metrics
A primary bottleneck under the IVDR is the severe imbalance between the demand for conformity assessments and the capacity of designated Notified Bodies. Under the previous Directive (IVDD), an estimated less than 10% of IVDs required NB intervention [57]. The IVDR's new risk-based classification system has inverted this, requiring approximately 80-90% of devices to undergo a mandatory third-party review [58] [57]. However, as of early 2025, only about 12-20 NBs are designated under the IVDR, a number that remains insufficient despite recent additions like DNV [59] [58] [57].
The 2025 EU Notified Bodies Survey quantifies the resulting strain. Data shows a significant backlog, with processing times extending between 13 to 18 months for the majority of applications [17] [58]. Critically, delays are not solely due to NB capacity. An EU Commission survey found that incomplete or poorly structured technical documentation from manufacturers accounts for approximately 58% of the total processing time [1]. This underscores a dual challenge: systemic capacity limits compounded by industry adaptation hurdles.
Table: Notified Body Capacity and Certification Metrics (2025 Data)
| Metric | Data | Source / Context |
|---|---|---|
| Designated IVDR Notified Bodies | ~12-20 | Number of NBs designated under IVDR as of early 2025 [58] [57]. |
| MDR Applications vs. Certificates | 28,489 applications / 12,177 certificates | Illustrates the broader MDR/IVDR system backlog [1]. |
| Typical Certification Timeline | 13-18 months | Duration for 60% of cases from application to final certificate [1] [58]. |
| Manufacturer-Caused Delay | ~58% of total processing time | Attributed to incomplete documentation submissions [1]. |
| IVDs Requiring NB Review | 80-90% | Under IVDR, up from <10% under the old IVDD [58] [57]. |
IVDR Transition Deadline Landscape and Strategic Implications
To avert a market crisis, the European Commission extended transition deadlines through Regulation (EU) 2024/1860. These extensions provide a graduated compliance runway but introduce a complex set of intermediate milestones that manufacturers must meet to qualify [60] [61].
The final compliance deadlines are staggered by risk class: Class D devices must comply by December 31, 2027; Class C by December 31, 2028; and Class B & A sterile by December 31, 2029 [60] [57]. To benefit from these extensions, manufacturers must have implemented an IVDR-compliant Quality Management System (QMS) by May 26, 2025 [60]. Furthermore, a critical imminent deadline is September 26, 2025, by which manufacturers of legacy IVDD-certified devices and up-classified Class D devices must have signed a written agreement with an IVDR Notified Body [61]. Missing this milestone jeopardizes the ability to use the extended transition periods.
Table: Key IVDR Transition Deadlines and Requirements
| Deadline | Risk Class Impact | Required Action |
|---|---|---|
| 26 May 2025 | All Legacy IVDs | Implement an IVDR-compliant Quality Management System (QMS) [60]. |
| 26 Sep 2025 | Legacy IVDD-certified & Class D | Sign a written agreement with an IVDR Notified Body [61]. |
| 26 May 2027 | Class B & A sterile | Submit a formal application to a Notified Body [60]. |
| 31 Dec 2027 | Class D | Full IVDR compliance deadline [60] [57]. |
| 31 Dec 2028 | Class C | Full IVDR compliance deadline [60] [57]. |
| 31 Dec 2029 | Class B & A sterile | Full IVDR compliance deadline [60] [57]. |
IVDR Transition Deadline Logic Map
Comparative Analysis: US FDA vs. EU IVDR Regulatory Pathways
The regulatory philosophies governing IVDs in the U.S. and EU have diverged significantly, creating distinct strategic landscapes. The U.S. FDA's approach is adaptive and risk-based, emphasizing total product lifecycle management and leveraging existing predicates through the 510(k) pathway [1] [56]. For AI-driven devices, the FDA's Predetermined Change Control Plan (PCCP) allows pre-approved iterative updates, fostering continuous innovation [1] [56].
Conversely, the EU IVDR establishes a prescriptive, precautionary framework. It requires a demonstration of conformity against absolute safety and performance standards, with limited recognition of equivalence to legacy devices [1]. The recent EU AI Act superimposes an additional layer of regulation for AI-based IVDs, classifying most as high-risk and demanding stringent governance, data quality, and transparency requirements [1]. This creates a dual regulatory burden (IVDR + AI Act) not present in the U.S. The impact is measurable: a 2025 survey indicated a 40% drop in the EU being selected as the first launch market for large IVD manufacturers since MDR/IVDR implementation [1].
Table: Comparison of US FDA and EU IVDR Regulatory Pathways
| Aspect | US FDA Pathway | EU IVDR Pathway |
|---|---|---|
| Core Philosophy | Risk-based, pro-innovation, lifecycle management [1] [56]. | Precautionary principle, high evidentiary burden for safety & performance [1]. |
| Key Mechanism | 510(k) (Substantial Equivalence), PCCP for AI [1] [56]. | Conformity Assessment by Notified Body, "exact equivalence" rarely accepted [1]. |
| AI/Software Integration | Encouraged via PCCP and Good Machine Learning Practice (GMLP) [1] [56]. | Stringent dual regulation under IVDR and the EU AI Act [1]. |
| Cybersecurity | Lifecycle approach via Secure Product Development Framework (SPDF) and SBOM [1]. | Requirements embedded in MDR/IVDR Annex I, plus NIS 2 Directive & Radio Equipment Directive [1]. |
| Strategic Impact | Favors the U.S. as a first-launch market; faster cycle for iterative innovation [1]. | Extended timelines, higher cost, and complexity delay EU market access [1] [57]. |
Experimental Protocols for IVDR Compliance Studies
For researchers generating clinical evidence for IVDR compliance, rigorous and predefined protocols are essential. The following methodologies are critical for performance evaluation.
Protocol for Clinical Performance Studies (IVDR Annex XIII)
This protocol generates evidence for clinical performance and scientific validity.
- Study Design: Define as a prospective, retrospective, or mixed study following ISO 20916 standards. Justify the choice based on target population availability and claim [57].
- Site & Sample Selection: Select clinical sites with appropriate patient populations. Specify inclusion/exclusion criteria and procedures for obtaining residual human samples with ethical approval and informed consent where applicable.
- Index Test Procedure: Detail the execution of the IVD under evaluation using predefined instructions for use. Incorporate blinding to the reference method result.
- Reference Standard Comparison: Employ a clinically accepted reference method (e.g., clinical diagnosis, well-established comparator assay). Document all procedures.
- Data Analysis Plan: Pre-specify statistical endpoints (e.g., sensitivity, specificity, PPV, NPV) with confidence intervals. Plan for subgroup analyses.
Protocol for Analytical Performance Testing
This protocol validates the analytical sensitivity, specificity, precision, and accuracy of the IVD.
- Test Matrix Definition: Identify all relevant sample types (e.g., serum, plasma, swab) and interfering substances to be tested.
- Experimental Replication: Design experiments with sufficient replicates (e.g., n=3 per run over 5 days) to robustly assess within-run, between-run, and total precision.
- Reference Material Use: Use standardized, traceable reference materials (e.g., WHO International Standards) for calibration and accuracy studies.
- Limit of Detection (LoD) & Quantification (LoQ): Determine LoD via serial dilution studies in relevant matrix. Establish LoQ as the lowest level meeting predefined precision (CV%) and bias criteria.
- Data Documentation: Record all raw data, instrument logs, and reagent lot numbers in a controlled, audit-ready format.
EU IVDR Notified Body Certification Workflow
Successfully navigating IVDR requirements demands a suite of specialized tools and resources. This toolkit outlines essential components for building a compliant technical dossier and quality system.
Table: Research Reagent Solutions & Essential Materials for IVDR Compliance
| Tool/Resource | Function in IVDR Compliance | Key Consideration |
|---|---|---|
| Certified Reference Materials | Provide traceable standards for analytical performance studies (accuracy, LoD) [62]. | Must be internationally recognized (e.g., WHO IS) for Class C/D devices. |
| Well-Characterized Biobank Samples | Serve as validated specimens for clinical performance studies [57]. | Ethical sourcing, informed consent, and relevant clinical data linkage are critical. |
| Quality Management System (QMS) Software | Manages document control, CAPA, audits, and training per Article 10(8) and ISO 13485 [60] [58]. | Should be validated for 21 CFR Part 11 / Annex 11 compliance for audit readiness. |
| Electronic Data Capture (EDC) System | Captures and manages clinical performance study data per ISO 20916 [57]. | Must ensure data integrity, audit trail, and compliance with GDPR for EU studies. |
| Unique Device Identification (UDI) Service | Generates and manages device identifiers for traceability (Article 24) [58]. | Requires integration with manufacturing and labeling processes. |
| Post-Market Surveillance (PMS) Platform | Systematically collects and analyzes post-market data (PMSR/PSUR) per Annex III [62] [58]. | Should facilitate trend reporting and signal detection from multiple sources. |
| Gap Assessment Template | Benchmarks existing technical documentation and processes against IVDR Annex II/III requirements [62] [58]. | A structured first step in any transition project to prioritize efforts. |
This guide provides a comparative analysis of the regulatory pathways for In Vitro Diagnostics (IVDs) in the United States and the European Union, with a specific focus on the recent evolution of policy for Laboratory Developed Tests (LDTs). The content is framed within a broader thesis on US-EU regulatory divergence, highlighting how differing philosophies—pro-innovation versus precautionary—shape compliance strategies for researchers and developers [1].
Current Regulatory Status of LDTs: In a significant recent development, the U.S. Food and Drug Administration's (FDA) 2024 final rule to actively regulate LDTs as medical devices has been vacated by a federal court and formally rescinded. As of September 19, 2025, the FDA has reverted to its prior policy of enforcement discretion for most LDTs, meaning they are not subject to FDA premarket review requirements [63] [64]. The court ruled that the FDA lacked the statutory authority under the Federal Food, Drug, and Cosmetic Act to regulate LDTs, which are considered a professional service, as medical devices [65]. Therefore, the phased compliance approach outlined in the 2024 rule is no longer in effect. The following analysis of the phased approach and the comparative regulatory landscape is presented for educational and strategic planning purposes, reflecting the frameworks that would have been applicable and that continue to govern commercially distributed IVDs.
Comparative Analysis of US and EU IVD Regulatory Pathways
The regulatory philosophies and structures governing IVDs in the US and EU create distinct pathways to market. The US framework is characterized by centralized FDA oversight with a focus on risk-based classification, while the EU's In Vitro Diagnostic Regulation (IVDR) establishes a more decentralized system with heightened requirements for clinical evidence and post-market surveillance [1] [3].
Table: Core Comparison of US FDA and EU IVD Regulatory Frameworks
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Federal Food, Drug & Cosmetic Act (FD&C Act); 21 CFR Parts 809, 820, 807 [3]. | In Vitro Diagnostic Regulation (EU) 2017/746 [3]. |
| Risk Classification | 3 Classes: Class I (lowest risk), Class II, Class III (highest risk). Based on intended use and risk [3]. | 4 Classes: Class A (lowest risk), B, C, D (highest risk). Rules-based per Annex VIII [3]. |
| Conformity Assessment Body | FDA (centralized agency). | Notified Body (NB) (private organization designated by an EU member state). |
| Premarket Authorization Pathway | Class I: Mostly exempt from premarket submission. Class II: Premarket Notification [510(k)] (demonstration of substantial equivalence). Class III: Premarket Approval (PMA) (scientific evidence of safety & effectiveness) [3]. | Class A (non-sterile): Self-declaration. Classes A (sterile) through D: Involvement of a Notified Body. Requires technical documentation review and, for Class C & D, assessment of performance evaluation [3] [28]. |
| Clinical Evidence Requirement | Required for Class III and some Class II devices. Clinical performance testing is required for all IVDs [3]. | Performance Evaluation required for all classes, consisting of Analytical Performance, Clinical Performance, and Scientific Validity. Evidence must be proportionate to risk class [3]. |
| Post-Market Surveillance (PMS) | Medical Device Reporting (MDR) for adverse events. No mandatory periodic reporting for all classes [3]. | Required for all classes. Periodic Safety Update Report (PSUR) for Class C & D; Post-Market Surveillance Report (PMSR) for Class A & B [3]. |
| Unique Device Identification (UDI) | Required [3]. | Required, with data stored in the European Database on Medical Devices (EUDAMED) [3]. |
The Phased LDT Rule (Historical Context): The now-rescinded 2024 FDA Final Rule proposed a five-stage, four-year phased approach to end enforcement discretion [66]. This guide will reference this structure as a basis for comparing regulatory rigor against the EU's IVDR framework for in-house devices.
Table: Phased Compliance Approach of the FDA's 2024 LDT Final Rule (Rescinded)
| Stage | Deadline | Core Requirements |
|---|---|---|
| Stage 1 | May 6, 2025 | Medical Device Reporting (MDR), Correction & Removal Reporting, Complaint Files [66]. |
| Stage 2 | May 6, 2026 | Establishment Registration & Listing, Labeling, Investigational Use [66]. |
| Stage 3 | May 6, 2027 | Quality System Regulation (QSR) / Quality Management System (QMS) requirements [66]. |
| Stage 4 | Nov 6, 2027 | Premarket Approval (PMA) submissions for high-risk (Class III) LDTs [66]. |
| Stage 5 | May 6, 2028 | 510(k) or De Novo submissions for moderate- and low-risk LDTs [66]. |
The divergence extends to strategic implications. Data indicates a shift toward a "US-First" launch model for innovative MedTech, attributed to the more predictable US pathway and the perceived complexity of the IVDR [1]. Since the implementation of the EU's regulations, the choice of the EU as a first launch market has decreased significantly for large manufacturers [1].
Regulatory Pathways for IVDs in the US and EU
Experimental Protocols and Evidence Generation
The core of IVD compliance lies in generating robust analytical and clinical performance data. While the US and EU require similar fundamental scientific evidence, the structure, rigor, and documentation expectations differ, particularly under the IVDR's Performance Evaluation requirement [3].
Analytical Performance Validation Protocol
This foundational protocol evaluates the test's technical performance characteristics. The data generated is essential for both US submissions (for IVDs) and the Analytical Performance pillar of the EU's Performance Evaluation [3].
Objective: To rigorously determine and document the analytical characteristics of an IVD/LDT, including its sensitivity, specificity, precision, accuracy, and reportable range.
Experimental Design & Methodology:
- Sample Selection: Use well-characterized clinical samples or standardized reference materials. Include samples spanning the assay's measurable range and critical medical decision points. For infectious disease tests, include panels with known concentrations of target pathogen [67].
- Precision (Repeatability & Reproducibility):
- Intra-assay Precision: Run 20 replicates of 2-3 samples (low, mid, high concentration) in a single run by one operator.
- Inter-assay Precision: Run 2-3 samples in duplicate across 20 different runs, over 10-20 days, using different lots of reagents and calibrators if applicable.
- Calculation: Determine mean, standard deviation (SD), and coefficient of variation (%CV) for quantitative assays. For qualitative assays, report percent agreement.
- Analytical Sensitivity (Limit of Detection - LoD):
- Prepare a dilution series of the target analyte in the appropriate matrix.
- Test a minimum of 20 replicates per dilution near the expected LoD.
- The LoD is the lowest concentration at which ≥95% of replicates are detected.
- Analytical Specificity/Interference:
- Cross-reactivity: Test against a panel of related pathogens or structurally similar molecules.
- Interfering Substances: Spike samples with common interferents (e.g., hemoglobin, lipids, bilirubin, common medications) at clinically relevant concentrations and assess recovery.
- Reportable Range/Linearity: Test a series of samples with analyte concentrations across the claimed measuring range. Perform polynomial regression analysis; linearity is accepted if the deviation from the fitted line is within pre-defined acceptance criteria.
Data Analysis & Acceptance Criteria: Predefined acceptance criteria must be based on intended use, clinical requirements, and state-of-the-art. For example, precision %CV should be <15% for most biomarkers. All protocols must be documented in a validation plan, and results summarized in a report suitable for regulatory submission [67].
Clinical Performance Study Protocol
This protocol assesses the test's ability to correctly identify a clinical condition or status in a target population. It forms the basis for the Clinical Performance and Scientific Validity pillars of the EU IVDR Performance Evaluation and is critical for US PMA and many 510(k) submissions [3] [68].
Objective: To estimate the clinical sensitivity, specificity, and predictive values of the IVD in an intended use population.
Experimental Design & Methodology:
- Study Design: A prospective, observational study is preferred. Retrospective studies using archived samples with well-defined clinical data are also acceptable but require justification.
- Subject/Sample Selection:
- Case Subjects: A cohort with the target condition, confirmed by a reference standard method (e.g., clinical diagnosis, gold standard test, imaging).
- Control Subjects: A cohort without the target condition, representing the population in which the test will be used. This may include healthy individuals and those with conditions that are differential diagnoses.
- Sample Size: Must be statistically justified to provide precise estimates (e.g., confidence intervals) for sensitivity and specificity. A minimum of 100 positive and 100 negative samples is often a starting point for feasibility.
- Blinding: The personnel performing the index test (the new IVD) should be blinded to the reference standard result, and vice versa, to prevent bias.
- Testing Procedure: Execute the test according to its finalized instructions for use (IFU). Document all procedural deviations.
- Reference Standard: Clearly define and justify the method used to establish the "true" disease status. This is often the greatest source of challenge in clinical validation [68].
Data Analysis:
- Construct a 2x2 contingency table comparing index test results against the reference standard.
- Calculate: Clinical Sensitivity, Specificity, Positive Predictive Value (PPV), Negative Predictive Value (NPV), and their 95% confidence intervals.
- For quantitative tests, generate a Receiver Operating Characteristic (ROC) curve and determine the optimal clinical cut-off value.
Regulatory Integration: Under the EU IVDR, this study may be a performance study requiring notification or authorization from competent authorities, especially if it is interventional [68]. In the US, such studies are reviewed as part of the premarket submission. For LDTs used in clinical trials of therapeutics, early engagement with both FDA (for the drug) and EU authorities (for the IVDR status of the assay) is critical to avoid delays [68].
Table: Comparison of Evidence Requirements for a High-Risk (US Class III / EU Class D) IVD
| Evidence Component | US FDA (PMA) | EU IVDR (Class D) | Key Differences in Emphasis |
|---|---|---|---|
| Analytical Performance | Extensive data required. Follows FDA guidance and recognized standards [67] [3]. | Pillar of Performance Evaluation. Must meet General Safety and Performance Requirements (GSPRs) [3]. | Largely aligned technically. EU requires explicit linkage to GSPRs in documentation. |
| Clinical Performance | Data from a pivotal clinical study required to demonstrate safety & effectiveness [3]. | Pillar of Performance Evaluation. Requires scientific validity and clinical performance data [3]. | EU requires a more structured Performance Evaluation Report integrating all evidence. US focuses on a single pivotal study. |
| Post-Market Plan | Required as part of submission. May include commitments for post-approval studies [3]. | Post-Market Performance Follow-up (PMPF) Plan required. PMPF actively updates the Performance Evaluation [3]. | EU PMPF is a dynamic, continuous process explicitly tied to the device's lifecycle evidence. US post-approval studies are often discrete. |
| Literature Review | Can be used to support certain claims. | Mandatory for Scientific Validity. Must be a systematic literature review [3]. | The systematic nature is a formal, mandated requirement under IVDR. |
Workflow for IVD Performance Evidence Generation
The Scientist's Toolkit: Research Reagent Solutions
Selecting appropriate materials is critical for generating defensible regulatory data. The following table details key reagents and their functions in IVD development and validation [67].
Table: Essential Research Reagents for IVD Development & Validation
| Reagent/Material | Function in Development/Validation | Critical Considerations for Compliance |
|---|---|---|
| Certified Reference Materials (CRMs) | Provide an analyte value traceable to an international standard. Used for calibrator assignment, accuracy assessment, and trueness determination. | Source from recognized bodies (e.g., NIST, IRMM). Documentation of certificate of analysis and traceability is essential for technical file [67]. |
| Characterized Clinical Samples | Form the basis for clinical sensitivity/specificity studies. Used for precision, interference, and LoD experiments. | Must be well-characterized using a reference method. Ethical acquisition and informed consent are mandatory. Documentation of sample provenance, storage conditions, and stability is required [68]. |
| Third-Party Contrived Controls & Panels | Evaluate analytical specificity (cross-reactivity) and detectability of rare strains/variants. Assess assay performance against a broad microbial or genetic panel. | Panels should be clinically relevant and challenge the assay's claims. Vendor reports on panel composition and characterization support the data [67]. |
| Interferent Stocks (e.g., Hemoglobin, Lipids, Bilirubin) | Used in interference studies per CLSI EP07 guideline to demonstrate assay robustness against common sample matrix interferents. | Use purified substances at high concentrations. Spiking studies must use clinically relevant concentrations and appropriate controls [67]. |
| Molecular Grade Enzymes & Master Mixes | Core components for molecular-based LDTs/IVDs (e.g., PCR, NGS). Determine assay efficiency, sensitivity, and specificity. | Select reagents with documentation of purity, absence of contaminating nucleases, and consistent lot-to-lot performance. Validation must account for reagent lot changes [67]. |
Strategies for Legacy Devices and "Grandfathering" in Both Regions
The global in vitro diagnostic (IVD) market is characterized by a profound and widening regulatory divide between the United States (US) and the European Union (EU). This divergence creates distinct strategic imperatives for IVD manufacturers, particularly concerning legacy devices and the policies governing their continued market access. In the US, a pro-innovation stance facilitates iterative development and market entry, while the EU’s precautionary principle has established a more complex and stringent environment [1]. Understanding the “grandfathering” provisions and transition strategies for existing products is crucial for efficient portfolio management and global commercialization.
This guide objectively compares the performance of different regulatory strategies for legacy IVDs in both regions, framed within a broader thesis on US versus EU regulatory requirements. The analysis is intended for researchers, scientists, and drug development professionals who must navigate these landscapes to bring diagnostic innovations to patients.
Comparative Analysis of US and EU Regulatory Frameworks
The foundational philosophies of the US Food and Drug Administration (FDA) and the European system under the In Vitro Diagnostic Regulation (IVDR) differ significantly, influencing all aspects of device approval and lifecycle management.
US Framework (FDA): The US system is centralized under the FDA and employs a risk-based classification (Class I, II, III) [3]. Its most notable feature for legacy devices is the 510(k) substantial equivalence pathway. This allows a new device to leverage the regulatory history of a legally marketed predicate device, facilitating a more streamlined review process [6] [69]. For true legacy devices, the US operates on a "grandfathering" principle for devices on the market prior to May 28, 1976, which are not subject to premarket approval (PMA) requirements if their design and intended use remain unchanged [3].
EU Framework (IVDR): The EU system is decentralized, relying on independent Notified Bodies (NBs) designated by member states to assess conformity [1] [69]. The IVDR introduced a more rigorous, rule-based classification system with four risk classes (A through D) [3]. A critical challenge under the IVDR is its limited “grandfathering.” The regulation estimates that 80-90% of IVDs previously under the Directive now require Notified Body review, a massive shift from the prior regime [3]. To avert a market crisis, the EU extended transition deadlines: Class D devices must comply by December 2027, Class C by December 2028, and Class A & B by December 2029 [1].
Table: Comparative Overview of US and EU IVD Regulatory Frameworks
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Food, Drug & Cosmetic Act; 21 CFR Parts 809, 812, 820 [3] | Regulation (EU) 2017/746 [3] |
| Classification | 3 Classes (I, II, III) [3] | 4 Classes (A, B, C, D) [3] |
| Key Market Pathway | 510(k) (Substantial Equivalence), PMA, De Novo [6] [69] | Conformity Assessment via Notified Body (Annexes IX-XI) [69] |
| "Grandfathering" Policy | Exists for pre-1976 devices; heavy reliance on predicate-based 510(k) [3] | Very limited; extended transition periods for legacy devices until 2027-2029 [1] |
| Clinical Evidence | Required for Class III and some Class II; focus on analytical and clinical performance [3] | Performance Evaluation required for all classes (Analytical, Clinical, Scientific Validity) [3] |
| Post-Market Surveillance | Medical Device Reporting (MDR), Annual Reports (PMA) [6] | Periodic Safety Update Report (PSUR) for Class C/D, Post-Market Surveillance Report (PMSR) for Class A/B [3] |
| Quality System | Quality System Regulation (QSR)/transitioning to ISO 13485 alignment by 2026 [6] | ISO 13485:2016 required [6] |
Market Entry Strategy and Performance Data
The choice between a US-first or EU-first market entry strategy has become a critical commercial decision. Data indicates a strong trend toward launching first in the US due to predictable pathways and faster access.
Timelines and Costs: An analysis of market entry pathways reveals a consistent performance advantage for the US 510(k) route in terms of speed. The FDA 510(k) process averages 6-12 months from submission to clearance. In contrast, achieving CE marking under the IVDR typically requires 12-18 months for Notified Body review and certification [6]. Financially, the IVDR process is estimated to cost between $500,000 and $2 million, while a US 510(k) can range from $1 million to $6 million, though the latter figure often includes more substantial clinical study costs [6].
Strategic Outcomes: This divergence has measurable impacts on market behavior. Since the implementation of the MDR/IVDR, the choice of the EU as the first launch market has dropped by approximately 40% for large IVD manufacturers [1]. The US now accounts for about 46.4% of the global MedTech market, compared to Europe's 26.4% [1]. The "US-First" model allows companies to generate early revenue and gather real-world evidence that can later support the more evidence-intensive EU submission [1].
Table: Comparison of Market Entry Strategy Performance
| Metric | US-First Strategy (via 510(k)) | EU-First Strategy (via IVDR) | Notes |
|---|---|---|---|
| Average Timeline | 6-12 months [6] | 12-18 months [6] | EU timeline heavily depends on Notified Body capacity and application quality. |
| Estimated Direct Cost | $1M - $6M [6] | $500K - $2M [6] | US costs are higher largely due to clinical study expenses; EU costs focus on NB fees and documentation. |
| Time to Revenue | Faster | Slower | Earlier US launch accelerates commercial return on investment. |
| Evidence for 2nd Market | Generates RWE for EU CER | Strong clinical data supports US PMA or De Novo | EU's rigorous Performance Evaluation aids a subsequent US submission for novel devices. |
| Market Impact | Preferred for incremental innovation & speed [1] | Necessary for EU access and global expansion credibility | EU approval (CE Mark) facilitates entry into other international markets. |
Experimental and Clinical Validation Protocols
Successful regulatory submission in either region hinges on robust experimental data. The protocols for generating this evidence, while sharing common scientific principles, are shaped by different regulatory expectations.
Analytical Performance Validation: This foundational protocol tests the intrinsic technical capabilities of the IVD.
- Objective: To precisely measure accuracy, precision, sensitivity, specificity, reportable range, and limits of detection/quantitation.
- Protocol: Test a defined number of replicates (e.g., n=20) across multiple days using clinical samples and/or reference materials that span the assay's measuring range. For EU submissions, particular emphasis is placed on using samples representative of the intended use population in terms of demographics and medical conditions [70].
- Key Difference: The IVDR's General Safety and Performance Requirements (GSPRs) require a more explicitly documented link between the validation plan, the risk analysis, and the stated performance claims.
Clinical Performance Study (Diagnostic Accuracy): This protocol evaluates the device's ability to correctly identify a clinical condition.
- Objective: To determine clinical sensitivity and specificity versus a clinical reference standard (e.g., diagnostic biopsy, physician diagnosis).
- Protocol: Conduct a prospective or retrospective study using well-characterized clinical samples from intended-use patients and controls. Sample size must be statistically justified to achieve adequate confidence intervals. For the EU Performance Evaluation, this study is one of three pillars and must be integrated with data on scientific validity and analytical performance [3].
- Key Difference: The FDA may accept a predicate comparison for 510(k) submissions, while the IVDR demands a direct clinical evaluation for the specific device, with equivalence claims to other devices being very difficult to substantiate [6].
AI/Software-Enabled Device Validation: For IVDs incorporating artificial intelligence/machine learning (AI/ML), additional protocols are required.
- Objective: To demonstrate algorithm robustness, freedom from unacceptable bias, and stability across diverse data sets.
- Protocol: Use a locked algorithm to analyze a completely independent validation dataset not used in training or tuning. Perform extensive stress-testing with edge cases and demographic sub-groups to identify bias [70]. In the US, the Predetermined Change Control Plan (PCCP) guidance allows for a roadmap for future iterative algorithm changes [1]. The EU AI Act imposes explicit requirements for data governance, human oversight, and transparency of the AI system's logic and limitations [1] [70].
Regulatory Pathway Comparison for IVDs
The Scientist's Toolkit: Essential Research Reagent Solutions
Building a compelling regulatory submission requires meticulous work with high-quality materials. Below is a table of essential research reagents and their functions in the development and validation of IVDs.
Table: Key Research Reagent Solutions for IVD Development & Validation
| Reagent/Material | Primary Function | Regulatory Significance |
|---|---|---|
| Certified Reference Materials (CRMs) | Provide a metrological traceability chain to an international standard; used to calibrate instruments and validate assay accuracy. | Critical for demonstrating analytical accuracy in both US and EU submissions. Essential for IVDR compliance with metrological traceability requirements. |
| Characterized Clinical Sample Panels | Well-defined patient samples (positive/negative) used to establish clinical sensitivity, specificity, and predictive values. | The foundation of the clinical performance study. Diversity of panels (age, gender, ethnicity, disease stage) is crucial, especially under EU guidance on representativeness [70]. |
| Interferent/Cross-Reactivity Panels | Substances (e.g., lipids, bilirubin, common drugs, biotin) or structurally similar analytes tested to assess assay specificity. | Directly addresses risk management and analytical specificity requirements. Documentation is a key part of the technical file. |
| Quality Control Materials | Stable materials with known target values run daily to monitor assay precision and long-term performance stability. | Required for both pre-market validation and ongoing post-market surveillance. Data supports assay robustness and is part of the post-market performance follow-up (PMPF) in the EU. |
| Proteomic/Genomic Standards | For molecular or protein-based assays, these provide standardized fragments, sequences, or proteins for assay optimization and reproducibility testing. | Ensures consistency in development and validation phases. Important for demonstrating analytical sensitivity (Limit of Detection). |
| Software Validation Suite | For digital/ALML-enabled IVDs, a comprehensive set of input data used to test algorithm performance, including edge cases and adversarial examples. | Core to establishing algorithm robustness and freedom from bias. Mandatory for compliance with FDA PCCP and EU AI Act principles [1] [70]. |
IVDR Performance Evaluation Workflow
Post-Market Surveillance (PMS) and Vigilance Reporting Obligations
This comparison guide objectively evaluates the performance of the European Union (EU) and United States (US) regulatory frameworks for post-market surveillance (PMS) and vigilance of In Vitro Diagnostic (IVD) devices. Framed within broader research on US versus EU IVD regulations, the analysis is supported by recent experimental data on incident reporting trends and the effectiveness of corrective actions. The findings are intended to inform researchers, scientists, and drug development professionals navigating global compliance.
The EU and US systems share the fundamental objective of monitoring device safety but differ significantly in structure, proactive requirements, and data transparency. The EU's In Vitro Diagnostic Regulation (IVDR) mandates a proactive, planned, and continuous PMS system integrated with periodic reporting for all device classes [3] [71]. In contrast, the US FDA's framework is more reactive, centered on mandatory adverse event reporting, with proactive surveillance studies required only for a subset of higher-risk devices [72]. Recent data indicates that while both systems capture similar primary failure modes, the structured EU approach may facilitate more systematic trend analysis and earlier signal detection [73].
Key Performance Indicators (KPIs) at a Glance:
| Performance Metric | EU IVDR System | US FDA System | Experimental Data Insight [73] |
|---|---|---|---|
| System Architecture | Proactive, plan-based for all devices [3]. | Reactive, report-triggered for all; proactive studies for select devices [72]. | Proactive systems correlate with more consistent FSCA implementation. |
| Reporting Timeline (Serious Incidents) | 15 days for serious public health threat; 30 days for other serious incidents [3]. | 30 days for serious injury/unanticipated death; 5 days for catastrophic events [72]. | Timelines affect speed of risk mitigation; regional variations exist. |
| Primary Analytical Output | Periodic Safety Update Report (PSUR) for Class C/D; Post-Market Surveillance Report for Class A/B [3] [71]. | Annual PMS Report for Section 522 studies; No equivalent to PSUR for most devices [72]. | PSURs provide structured benefit-risk reassessment. |
| Central Database | EUDAMED (mandatory for actor/device registration, incident reporting) [74] [23]. | MAUDE (public adverse event database) [73]. | EUDAMED aims for higher structured data integrity; MAUDE offers broader public access. |
| Coverage of IVDs | All IVD classes (A-D) have explicit PMS obligations [3]. | Focus on Class II/III; many Class I/510(k)-exempt devices have minimal requirements [10] [72]. | Broader coverage captures more data across device spectrum. |
Detailed Comparison of Regulatory Performance
The following tables break down the core components, obligations, and outputs of each system, providing a basis for direct comparison.
Table 1: Foundation and Core Obligations
| Regulatory Component | EU IVDR Framework | US FDA Framework |
|---|---|---|
| Legal Basis | Regulation (EU) 2017/746 (IVDR), fully enforceable [3]. | Federal Food, Drug & Cosmetic Act; 21 CFR Parts 803, 806, 822 [72]. |
| Scope of PMS | Comprehensive & Continuous: Required for all IVDs. Integrated into QMS and life cycle [3]. | Conditional & Reactive: MDR required for all. Proactive Sec. 522 studies only for designated high-risk devices [72]. |
| System Requirement | Mandatory PMS Plan for each device, proportionate to risk class [3] [71]. | No general PMS plan requirement. A Surveillance Plan is required only for devices subject to a Section 522 order [72]. |
| Vigilance Reporting Trigger | Any serious incident involving a device [74]. | Reportable event: Death, serious injury, or malfunction likely to cause death/serious injury [72]. |
Table 2: Reporting Outputs and Documentation
| Output Document | EU IVDR Requirement | US FDA Requirement | Primary Function |
|---|---|---|---|
| Periodic Safety Update Report (PSUR) | Required for Class C & D devices. Summarizes PMS data, benefit-risk analysis, and CAPA status [3] [71]. | Not required. | Periodic, holistic safety review for high-risk devices. |
| Post-Market Surveillance Report (PMSR) | Required for Class A & B devices [3] [71]. | Not required. | Summary of PMS data for lower-risk devices. |
| Post-Market Surveillance (Sec. 522) Report | Not applicable. | Required for devices under a Section 522 order. Submitted annually and as a final report [72]. | Reports results of an FDA-ordered, targeted study. |
| Summary of Safety and Performance (SSP) | Required for Class C & D devices, publicly available [3]. | Not required. | Public transparency document on safety and performance. |
Experimental Protocol: Analyzing PMS System Effectiveness
A 2024 observational study provides a methodology for comparing the operational performance of different regulatory PMS systems [73].
1. Objective: To analyze post-market incident trends for high-risk devices and evaluate the effectiveness of Field Safety Corrective Actions (FSCAs) across different regulatory jurisdictions [73].
2. Data Sources & Collection:
- Primary Databases: Incidents and FSCAs were extracted from:
- Device Focus: Class IIb and III medical devices (analogous to EU Class C/D and US Class III IVDs) [73].
- Key Variables: Device type, manufacturer, incident date, failure mode (e.g., hardware, software, calibration), type of FSCA (recall, update, modification) [73].
3. Analytical Workflow: 1. Data Aggregation: Incidents from all sources were compiled into a unified dataset for 2024. 2. Categorization: Each incident was categorized by failure mode and device category. 3. Trend Analysis: Frequency and severity of incidents were analyzed to identify dominant risk patterns. 4. Outcome Measurement: The recurrence rate of issues for specific device categories was tracked before and after the implementation of FSCAs to measure their effectiveness [73].
Experimental Workflow for PMS Data Analysis
Experimental Data Analysis: Incident Trends and FSCA Outcomes
The experimental study yielded quantitative findings on device failures and regulatory actions [73].
Table 3: Analysis of Dominant Failure Modes in High-Risk Devices (2024 Data) [73]
| Device Category | Number of Reported Issues (Sample) | Examples of Specific Failure Modes |
|---|---|---|
| Orthopaedic & Implantable Devices | 4 | Implant corrosion, premature wear of hip implants, material fragility [73]. |
| Cardiac Monitoring & Implantable Devices | 3 | Battery life reduction, blood pump malfunction, connection issues [73]. |
| Invasive & Diagnostic Devices | 4 | Catheter breakage, stapler misfiring, lens fogging [73]. |
| Software-Driven Devices (e.g., infusion pumps) | High recurrence rate | Software malfunctions, calibration drift, algorithmic errors [73]. |
Table 4: Distribution and Effectiveness of Field Safety Corrective Actions (FSCAs) [73]
| Type of FSCA | Proportion of Implemented Actions | Observed Effectiveness & Context |
|---|---|---|
| Field Modification | 46% (Largest proportion) | Most effective for tangible hardware failures; significantly reduced recurrence for issues like battery or mechanical faults [73]. |
| Software Update | 26% | Variable effectiveness. Addressed immediate bugs but showed higher rates of persistent or new issues in software-driven devices [73]. |
| Recall (Device Removal) | 22% | Highly effective but most disruptive. Used for critical, non-mitigable risks like faulty critical components [73]. |
| Safety Notice | 6% | Preventive measure to update user instructions; effectiveness depends on user compliance [73]. |
Key Finding from Data: Hardware failures were the most frequently reported issue. While FSCAs like field modifications were largely effective for these, software-related problems proved more persistent, suggesting regulatory frameworks need enhanced tools for managing digital health technologies [73].
Post-Market Incident Analysis and Regulatory Response Pathway
Table 5: Essential Research Reagents and Resources
| Item & Source | Function in PMS/Vigilance Research |
|---|---|
| EUDAMED Database (European Commission) [74] | The primary EU repository for structured data on device registration, certificates, serious incidents, and FSCAs. Crucial for analyzing EU-specific trends. |
| MAUDE Database (U.S. FDA) [73] | The FDA's publicly accessible database of medical device reports. Essential for studying adverse event patterns and manufacturer compliance in the US market. |
| ISO 13485:2016 Standard | Specifies requirements for a Quality Management System (QMS). The foundation for integrating PMS processes, CAPA, and management review. The US QMSR (effective 2026) incorporates it by reference [23] [72]. |
| ISO 14971:2019 Standard | The framework for risk management of medical devices. Guides the ongoing benefit-risk assessment required by PMS, linking incident data to risk control measures [72]. |
| IVDR (EU 2017/746) & FDA 21 CFR Regulations [10] [3] | The primary regulatory texts. Provide the definitive legal requirements for PMS plans, performance evaluation, PSURs (IVDR), and adverse event reporting (FDA). |
| Clinical Performance Study Data | Data generated per IVDR Annex XIII or FDA IDE/PMA requirements. Serves as the baseline for post-market performance follow-up (PMPF) and the analysis of real-world versus clinical trial performance [3]. |
Future Directions and Evolving Landscapes
Both regulatory landscapes are evolving, with a noticeable trend toward harmonization of proactive principles and the integration of digital tools.
- Regulatory Reliance and Harmonization: Initiatives like the International Medical Device Regulators Forum (IMDRF) are promoting reliance on audits and approvals from other jurisdictions [75]. The UK MHRA's 2025 consultation also proposes routes for recognizing approvals from "comparable regulator countries" [75] [23].
- Digital Transformation and Transparency: The mandatory rollout of EUDAMED's UDI and device registration modules by 2026 will increase data transparency [23]. Similarly, the US FDA is expanding active surveillance using real-world data from electronic health records and registries [72].
- Focus on Software and AI: The EU's AI Act will integrate conformity assessments for high-risk AI systems into the IVDR process [23]. Both regions are emphasizing robust cybersecurity and lifecycle management for Software as a Medical Device (SaMD) and AI/ML-enabled IVDs [23] [72].
- Simplification and Refinement: The European Commission is consulting on simplifying the MDR and IVDR, with a draft expected by end of 2025 [75]. The US FDA revoked the LDT Final Rule, re-aligning certain lab test regulations more closely with the EU's approach under IVDR Article 5(5) [75] [23].
Navigating the Convergence of IVDR and the EU AI Act for Software Workflows
The integration of Artificial Intelligence (AI) into in vitro diagnostic (IVD) software has created a complex dual regulatory framework in the European Union. AI-enabled IVD software must now comply simultaneously with the In Vitro Diagnostic Regulation (IVDR, EU 2017/746) and the Artificial Intelligence Act (AI Act, EU 2024/1689) [70] [76]. This convergence represents a pivotal development within the broader context of transatlantic regulatory research, highlighting a growing philosophical and procedural divide between the innovation-focused U.S. approach and the precautionary, rights-based EU model [77] [1].
This guide provides a comparative analysis of these overlapping requirements, offering researchers and developers a structured pathway for compliance. The guidance document MDCG 2025-6 is central to this analysis, as it clarifies the intended interplay between the two regulations and advocates for an integrated compliance strategy [78].
Comparative Analysis of IVDR and AI Act Requirements
For manufacturers of AI-based IVD software, the IVDR and the AI Act impose complementary but distinct obligations. The IVDR provides the foundational medical device requirements for safety and performance, while the AI Act introduces additional, AI-specific controls for risk, data, and transparency [70] [79]. The table below summarizes the key regulatory domains across both frameworks.
Table 1: Comparison of Key Requirements under the EU IVDR and the EU AI Act for AI-Enabled Software [70] [79] [78].
| Requirement | IVDR (EU 2017/746) | AI Act (EU 2024/1689) | Key Points of Convergence/Divergence |
|---|---|---|---|
| Scope & Classification | Applies to IVD medical devices, classified by risk (Class A, B, C, D). Rule-based system (Annex VIII). | Applies to all AI systems. An AI system is "high-risk" if it is a safety component of, or is itself, a product requiring a Notified Body under listed legislation (e.g., IVDR) [80] [81]. | AI components in IVD software requiring NB assessment are automatically classified as "high-risk" under the AI Act. The IVDR risk class remains unchanged [70]. |
| Quality Management System (QMS) | Requires a QMS per Annex IX, typically aligned with ISO 13485, covering safety and performance. | Providers must establish a QMS covering the entire AI lifecycle, with specific elements for data governance and human oversight (Article 17) [79]. | The AI Act adds AI-specific QMS elements. Manufacturers are encouraged to integrate these into their existing IVDR QMS [70] [78]. |
| Risk Management | Continuous risk management per ISO 14971, focused on patient safety and diagnostic performance. | Requires AI-specific risk management addressing unique risks like bias, robustness, and impacts on fundamental rights (Article 9) [79]. | IVDR focuses on clinical safety; the AI Act expands the scope to include ethical and societal risks. Processes should be integrated [70]. |
| Data Governance | Requires appropriate, representative data for performance evaluation, with GDPR compliance. | Imposes stricter requirements for training, validation, and testing datasets: representativeness, bias assessment, and detailed documentation (Article 10) [79]. | The AI Act introduces enhanced obligations for dataset quality, traceability, and bias mitigation beyond general IVDR data requirements [70] [78]. |
| Technical Documentation | Comprehensive technical documentation per Annexes II and III to demonstrate conformity with IVDR. | Requires detailed "technical documentation" for the AI system (Annex IV), covering design, development, training, and testing data [79]. | A single technical documentation file can be created to satisfy both regulations (AI Act Article 11(2)). The IVDR file should be expanded to include AI Annex IV data [78]. |
| Transparency & Human Oversight | Labeling and Instructions for Use (IFU) must ensure safe use. Human oversight is implied. | Explicit obligations for transparency, user awareness of AI interaction, explainability of outputs, and mechanisms for human oversight/override (Articles 13, 14) [76]. | The AI Act mandates specific, verifiable measures for explainability and human control, which must be reflected in the software design and IFU [70]. |
| Post-Market Surveillance (PMS) | Requires a PMS system to continuously monitor performance and safety (Articles 78-81). | Requires a post-market monitoring plan for high-risk AI systems, including monitoring for performance degradation and "model drift" [70]. | AI Act PMS requirements (e.g., algorithm monitoring) must be integrated into the existing IVDR PMS plan [70] [78]. |
| Substantial Modification | Changes affecting safety or performance require review and may trigger a new conformity assessment. | Defines "substantial modification" for AI systems (e.g., major retraining) and introduces the concept of a "predetermined change control plan" (PCCP) [70]. | The AI Act formalizes a PCCP approach, similar to the U.S. FDA's, allowing pre-approved modifications within a defined plan [70] [1]. |
| Conformity Assessment | Class B, C, and D devices require assessment by a Notified Body (NB). | High-risk AI systems require NB conformity assessment. Where applicable, the same NB should perform both assessments [70] [78]. | A single, combined conformity assessment procedure under the IVDR is intended, provided all AI Act requirements are integrated [78]. |
US FDA vs EU IVDR/AI Act: A Strategic Regulatory Comparison
The EU's integrated framework contrasts significantly with the U.S. approach, creating a strategic divide for global developers [77] [1]. The U.S. FDA regulates AI-based Software as a Medical Device (SaMD) under existing medical device law, employing a more flexible, risk-based pathway system without a standalone horizontal AI law [79].
Table 2: Comparison of U.S. FDA and EU (IVDR + AI Act) Regulatory Approaches for AI-IVD Software [77] [79] [1].
| Aspect | United States (FDA) | European Union (IVDR + AI Act) |
|---|---|---|
| Core Regulatory Philosophy | Pro-innovation, risk-based. Aims to balance safety with promoting technological advancement [1]. | Precautionary, rights-based. Prioritizes patient safety, fundamental rights, and rigorous ex-ante demonstration of safety and performance [1] [76]. |
| Governing Framework for AI | Sector-specific guidance under FDA authority. No comprehensive AI law. Utilizes existing pathways (510(k), De Novo, PMA) with adaptive policies like the Predetermined Change Control Plan (PCCP) [79] [1]. | Dual framework: Sector-specific IVDR + horizontal AI Act. Creates explicit, legally binding obligations for all high-risk AI systems [70] [79]. |
| Market Entry Pathway for Moderate-Risk AI-IVD | 510(k) clearance (most common). Demonstrates "substantial equivalence" to a predicate device. Clinical data may not always be required [77] [6]. | IVDR Conformity Assessment + AI Act Requirements. Requires a Notified Body audit. Clinical performance evaluation is always mandatory. No predicate-based equivalence for performance studies [77] [6]. |
| Approach to AI Software Changes | FDA PCCP Guidance. Allows manufacturers to pre-specify and gain approval for certain future modifications (e.g., algorithm retraining) within a validated plan, reducing submission burden [1]. | "Substantial modification" rules. Changes beyond a predefined scope trigger reassessment. The AI Act explicitly encourages PCCPs, but detailed EU guidance is under development [70] [78]. |
| Post-Market Evidence Generation | Can be used to support new indications or modifications post-clearance. | Post-Market Clinical Follow-up (PMCF) is a proactive, mandatory requirement under IVDR for most devices, requiring continuous clinical data collection [77]. |
| Transparency & Explainability | Encouraged through various guidance documents but generally less prescriptive than the EU. Focus is on analytical and clinical validation. | Legally mandated by the AI Act. Requires technical solutions for explainability and clear information to users, directly impacting trust and adoption [1] [76]. |
Experimental Protocols for Integrated Compliance
Navigating the convergent framework requires a methodical, integrated approach. The following protocols outline key experimental and procedural methodologies for achieving compliance.
Protocol: Establishing an Integrated Quality Management System (QMS)
Objective: To expand an existing ISO 13485-compliant QMS (for IVDR) to incorporate all AI Act-specific requirements [78]. Methodology:
- Gap Analysis: Map current QMS procedures against AI Act Article 17 requirements (e.g., data governance, record-keeping, human oversight design controls).
- Procedure Integration: Revise and augment existing SOPs rather than creating parallel systems.
- Example: Integrate AI dataset management and bias-checking protocols into the existing "Design and Development Planning" and "Design Validation" procedures [80].
- Audit Trail Enhancement: Ensure the QMS supports detailed, automatic logging of the AI system's development, training, and post-market performance as required by AI Act Article 12 [79].
- Notified Body Alignment: Engage with the chosen Notified Body early to confirm the integrated QMS approach meets their expectations for a combined assessment [70].
Protocol: Developing Unified Technical Documentation
Objective: To create a single technical documentation file that demonstrates conformity with both Annexes II/III of the IVDR and Annex IV of the AI Act [78]. Methodology:
- File Structure Expansion: Use the IVDR technical documentation structure as the backbone. Append new, dedicated sections for AI-specific Annex IV content [79].
- Critical AI Documentation: Ensure the following are comprehensively detailed:
- Data Governance Dossier: Description of training, validation, and testing datasets, including sources, demographics, annotation methods, and bias assessments [70] [79].
- Model Design & Rationale: Detailed system architecture, design choices, and the rationale behind algorithm selection [79].
- Validation for Robustness: Documentation of testing under diverse conditions and against adversarial inputs to demonstrate cybersecurity and robustness (AI Act Article 15) [78].
- Human Oversight Design: Description of the user interface, interpretability features, and override mechanisms implemented to fulfill AI Act Article 14 [76].
- Traceability: Maintain explicit traceability between risk management outputs, design specifications, validation results, and the instructions for use.
Protocol: Conducting an AI-Aware Performance Evaluation
Objective: To design and execute clinical performance studies that satisfy IVDR requirements while also generating evidence for AI Act mandates on accuracy, robustness, and bias mitigation. Methodology:
- Representative Dataset Curation: Actively recruit validation datasets that are representative of the intended use population and environment across factors like age, gender, ethnicity, geography, and disease prevalence (aligns with both IVDR and AI Act Article 10) [70] [79].
- Bias Detection & Analysis: Incorporate statistical analyses to detect performance disparities across relevant subpopulations. Document any identified biases and the mitigation strategies employed [79].
- Robustness Testing: Beyond standard clinical sensitivity/specificity, include testing for:
- Input Perturbations: Assessing performance with noisy or low-quality input data.
- Domain Shifts: Evaluating performance on data from slightly different clinical settings or equipment.
- Continuous Performance Monitoring Plan: Design the Post-Market Performance Follow-up (PMPF) plan to actively monitor for model drift and degradation in real-world use, as required by the AI Act's post-market monitoring provisions [70].
Successfully navigating the dual regulatory landscape requires specialized tools and resources. The following toolkit details essential solutions for researchers and development professionals.
Table 3: Research Reagent Solutions for AI-IVD Compliance and Development [1] [80].
| Tool/Resource Category | Specific Examples/Function | Primary Purpose in Compliance Workflow |
|---|---|---|
| QMS & Document Management | Electronic Quality Management System (eQMS) software with AI workflow modules. | Centralizes and controls integrated SOPs, technical documentation, and audit trails for combined IVDR/AI Act audits [80]. |
| Data Governance & Annotation | Secure, version-controlled data lake platforms; specialized medical data annotation tools with QA features. | Manages curated, documented datasets for training/validation, ensuring traceability and compliance with AI Act data governance rules [79]. |
| Algorithm Development & Bias Testing | MLops platforms (e.g., MLflow, Weights & Biases); open-source fairness audit libraries (e.g., AIF360, Fairlearn). | Tracks experiment lineage, model versions, and hyperparameters. Quantitatively assesses algorithmic bias across patient subgroups [79]. |
| Explainability & Transparency | Model interpretability libraries (e.g., SHAP, LIME); automated report generation tools. | Generates visual explanations for AI decisions to be integrated into clinical reports, supporting transparency requirements [76]. |
| Regulatory Intelligence | Subscription services to platforms like Emergo by UL, Greenlight Guru, or FDA/EMA/EU commission tracking tools. | Provides real-time updates on guidance (e.g., MDCG documents), standards, and regulatory changes in both the US and EU markets [78]. |
Visualizing the Compliance Workflow
The following diagram illustrates the integrated workflow for achieving compliance for an AI-enabled IVD software under the convergent EU IVDR and AI Act framework, based on guidance from MDCG 2025-6 [78].
Diagram: AI-IVDR Dual Compliance Workflow. This flowchart outlines the three-phase integrated process for achieving compliance for AI-enabled IVD software under the EU's IVDR and AI Act. The process begins with qualification and automatic high-risk classification, moves through integrated system development centered on a unified technical file, and culminates in a single conformity assessment followed by continuous lifecycle management [70] [78].
Strategic Validation and Comparative Analysis for Global Market Access
This guide provides a detailed, objective comparison of the regulatory frameworks for In Vitro Diagnostic (IVD) devices in the United States (US) and the European Union (EU). The analysis focuses on three critical pillars: the generation of clinical evidence, labeling requirements, and the implementation of the Unique Device Identification (UDI) system. For researchers, scientists, and drug development professionals, understanding these divergent pathways is essential for strategic global product development and compliance. The comparison is framed within ongoing research into US-EU regulatory convergence and divergence, highlighting how distinct approaches aim to achieve the common goal of ensuring device safety and performance.
Comparative Analysis of Regulatory Classification and Pathways
A fundamental difference between the US and EU systems lies in their risk classification structures and the associated market authorization pathways.
Table 1: Regulatory Classification and Market Pathways for IVDs
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Federal Food, Drug & Cosmetic Act; 21 CFR Parts 809, 812, 814 [10]. | In Vitro Diagnostic Regulation (IVDR) – (EU) 2017/746 [3]. |
| Risk Classification | 3 Classes (I, II, III). IVDs are regulated as medical devices [3] [10]. | 4 Classes (A, B, C, D), from lowest to highest risk, based on Annex VIII rules [3]. |
| Key Market Pathways | • 510(k) (Premarket Notification): For Class I/II devices demonstrating substantial equivalence to a predicate [10]. • De Novo: For novel, low-to-moderate risk devices with no predicate [10]. • Premarket Approval (PMA): For Class III devices [10]. | • Self-Certification: Only for Class A non-sterile devices [3]. • Notified Body Review: Required for all Class B, C, and D devices. Scrutiny intensity increases with class [3]. |
| "Grandfathering" / Legacy Devices | Devices legally marketed before May 28, 1976, or substantially equivalent to such a device, may not require a PMA [3]. | Under IVDR, most legacy devices (previously under IVDD) require Notified Body certification, significantly increasing oversight [3]. |
A key strategic divergence is the US concept of "substantial equivalence" to an existing predicate, which is central to the 510(k) pathway [10]. In contrast, the EU IVDR employs a rule-based classification system and mandates conformity assessment by a Notified Body for most device classes, with no general reliance on predicate devices [3].
Comparative Analysis of Clinical Evidence Requirements
The nature, timing, and documentation of clinical evidence differ markedly between the two regions.
Table 2: Clinical Evidence Requirements for IVDs
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Core Requirement | Clinical performance testing is required for all IVDs, but the extent of clinical evidence is tied to risk class and submission type [3]. | Clinical evidence is mandatory for all IVDs, derived from performance evaluation [3]. |
| Framework | Evidence is part of premarket submission (510(k), PMA). Focus is on analytical and clinical performance (sensitivity, specificity) vs. a predicate or truth standard [10]. | Performance Evaluation is a continuous process with three pillars: 1) Scientific Validity, 2) Analytical Performance, 3) Clinical Performance [3]. |
| Key Submission Documents | • Premarket Submission (510(k), PMA, De Novo) containing study reports.• Pre-Submission meetings are encouraged for complex devices [10]. | • Performance Evaluation Report (PER).• Periodic Safety Update Report (PSUR) for Class C & D devices [3]. |
| Post-Market Requirement | Postmarket surveillance studies may be ordered as a condition of approval [10]. | Post-Market Performance Follow-up (PMPF) is required to actively update the PER [3]. |
The EU's IVDR formalizes a more structured and continuous lifecycle approach to clinical evidence through the Performance Evaluation and PMPF, whereas the US system integrates clinical validation into the premarket submission with potential post-market conditions [3] [10].
Experimental Protocol 1: US Clinical Validation for a Novel Class II IVD (De Novo Pathway)
- Objective: To establish analytical and clinical performance for a novel biomarker assay where no predicate device exists.
- Methodology:
- Pre-Submission: Engage with FDA via a Q-Submission Pre-Sub meeting to agree on study design and endpoints [10].
- Analytical Performance Studies: Conduct studies per FDA guidance to establish precision, accuracy, sensitivity, specificity, reportable range, and reference intervals.
- Clinical Validation Study:
- Design: Retrospective or prospective case-control or cohort study.
- Sample Size: Justified by statistical hypotheses for clinical sensitivity and specificity.
- Comparator: Use a clinically accepted truth standard (e.g., clinician diagnosis, established diagnostic algorithm), not another device.
- Sites: Multiple clinical sites to ensure population diversity.
- Endpoints: Primary endpoints are clinical sensitivity and specificity with two-sided 95% confidence intervals.
- Data Analysis: Perform pre-specified statistical analyses. All data is compiled into a De Novo Classification Request submission [10].
Experimental Protocol 2: EU Performance Evaluation for a Class C IVD
- Objective: To generate and document the scientific validity, analytical performance, and clinical performance for a Class C device under IVDR.
- Methodology:
- Plan: Develop a Performance Evaluation Plan (PEP) detailing the strategy for addressing all three pillars.
- Scientific Validity: Conduct a literature review to establish the association between the analyte and the clinical condition.
- Analytical Performance: Perform studies per Common Specifications or state-of-the-art to verify manufacturer's claims.
- Clinical Performance:
- Sources: May use existing clinical data, results from equivalence studies to a legally marketed device, or new clinical performance studies.
- Study Design: If a new study is needed, design follows EN/ISO 20916 and is approved by an Ethics Committee.
- Analysis: Data is analyzed to establish performance metrics.
- Report & Update: Compile all data into a Performance Evaluation Report (PER). Initiate a PMPF plan to collect post-market data and update the PER annually [3].
Comparative Analysis of Labeling Requirements
Both regions have detailed labeling mandates to ensure safe and effective use, but their structures and emphases vary.
Table 3: Core Labeling Requirements for IVDs
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Text | 21 CFR Part 801 [82] and Part 809 (specific to IVDs) [3] [10]. | Annex I, Chapter III of IVDR ((EU) 2017/746). |
| Core Principles | Labeling must provide "adequate directions for use" so the layperson can use the device safely [82]. Information must be prominent and not misleading [82]. | Labeling must enable safe use and reflect the General Safety and Performance Requirements (GSPRs). Information must be clear to the intended user. |
| Language | English. | Official language(s) of the Member State where the device is sold. |
| Unique Device Identifier (UDI) | Must be presented in both human-readable (HRI) and machine-readable (AIDC) formats on the label and higher packaging [83] [84]. | Must be presented in both human-readable (HRI) and machine-readable (AIDC) formats on the label and all higher levels of packaging [85]. |
| Special Requirement | Compliant labeling is a prerequisite for obtaining market authorization (clearance/approval) [10]. | The Basic UDI-DI (a database key for device groups) must be included in technical documentation and certificates, but not on the physical label [85] [86]. |
Comparative Analysis of the Unique Device Identification (UDI) System
While both systems share a common goal of traceability and are based on international principles, their implementation details differ significantly.
Table 4: UDI System Implementation: US vs. EU
| Aspect | United States (FDA) | European Union (IVDR/MDR) |
|---|---|---|
| Database | Global UDI Database (GUDID). Publicly accessible via AccessGUDID [87] [83]. | European Database on Medical Devices (EUDAMED) (when fully operational) [85] [86]. |
| Data Submission Format | HL7 Structured Product Labeling (SPL), primarily via the Electronic Submissions Gateway [87] [86]. | XML format [86]. |
| Key Identifier | UDI-DI serves as the primary access key in the GUDID [83] [86]. | Basic UDI-DI is the primary access key in EUDAMED for a device group. The UDI-DI identifies a specific device model [85] [86]. |
| Nomenclature | Global Medical Device Nomenclature (GMDN) recommended. | European Medical Device Nomenclature (EMDN) mandatory [86]. |
| Compliance Dates (IVD Examples) | • Class III: Sep 24, 2016• Class II: Sep 24, 2018• Class I: Sep 24, 2020 [86]. | • Class D: May 26, 2023• Class C: May 26, 2025 [88]• Class B: May 26, 2025 [88]• Class A: May 26, 2027 [88]. |
A critical structural difference is the EU's introduction of the Basic UDI-DI, which groups device models for regulatory documentation, a concept not present in the US system [85] [86]. Furthermore, while the US GUDID is operational, the full mandatory use of EUDAMED's UDI module is pending its complete functionality [88] [86].
The Scientist's Toolkit: Essential Reagents & Materials for IVD Development
This table details key reagents and materials critical for generating the analytical and clinical performance data required for regulatory submissions in both regions.
Table 5: Key Research Reagent Solutions for IVD Development
| Item | Function in IVD Development | Regulatory Context |
|---|---|---|
| Analyte Specific Reagents (ASRs) | Antibodies, nucleic acid sequences, or ligands that specifically bind to a target analyte. They form the core detection mechanism of the IVD [10]. | In the US, ASRs are regulated as Class I, II, or III medical devices. Their sale and use are restricted [10]. |
| General Purpose Reagents (GPRs) | Chemical reagents (e.g., buffers, salts, enzymes) with general laboratory application for specimen preparation and examination, not specific to a single test [10]. | In the US, GPRs are typically Class I medical devices [10]. |
| Clinical Specimens (Biobanked) | Well-characterized human samples (serum, plasma, tissue) used to establish analytical specificity/sensitivity and initial clinical performance [10]. | Their use must be covered under an IRB/Ethics approval. Characterization (truth data) is critical for study validity. |
| Reference Materials & Standards | Materials with a defined quantity of an analyte used to calibrate equipment and validate method accuracy (trueness) [10]. | Use of internationally recognized standards (e.g., WHO International Standards) strengthens the validity of performance claims. |
| Control Materials | Materials used to monitor assay performance during validation and routine use (Positive, Negative, Quality Controls). | Their commutability with patient samples must be demonstrated. Manufacturing controls must follow Quality System Regulations [10]. |
For in vitro diagnostic (IVD) manufacturers, simultaneous access to the United States and European Union markets is not merely a commercial ambition but a strategic necessity. These two regions collectively represent a dominant share of the global MedTech market, yet their regulatory philosophies and pathways are increasingly divergent [1]. This analysis provides a structured comparison of the US FDA and EU IVDR frameworks, focusing on the empirical data, experimental protocols, and resource implications critical for researchers and drug development professionals. The goal is to inform a cost-benefit model for efficient resource allocation, enabling strategic decision-making in a complex global landscape. The European IVD market, valued at USD 23.35 billion in 2024 and projected to reach USD 34.47 billion by 2033, underscores the high stakes of successful market entry [29].
Comparative Regulatory Frameworks: US FDA vs. EU IVDR
A foundational understanding of the distinct regulatory classifications and requirements is essential for planning. The systems are both risk-based but apply different structures and levels of scrutiny.
Classification and Approval Pathways
The regulatory journey begins with classifying the device, which dictates the approval pathway, documentation, and timeline.
United States (FDA): The FDA classifies IVDs as medical devices into three categories: Class I (low risk), Class II (moderate risk), and Class III (high risk) [3] [10]. Most novel, high-risk devices require a Premarket Approval (PMA), which demands comprehensive clinical data to demonstrate safety and effectiveness. For devices substantially equivalent to a legally marketed predicate, the 510(k) premarket notification pathway is common [10]. The FDA also encourages a Pre-Submission process, allowing manufacturers to obtain formal feedback on study design or regulatory strategy before submitting a marketing application, which can reduce development costs and facilitate review [10].
European Union (IVDR): Under the In Vitro Diagnostic Regulation (IVDR 2017/746), IVDs are categorized into four risk classes: A (lowest), B, C, and D (highest) [3]. A pivotal change from the previous directive is that most devices now require a conformity assessment by a Notified Body (NB) [3] [89]. Unlike the US system, the IVDR eliminated "grandfathering" provisions for existing devices, requiring even well-established tests to undergo renewed scrutiny under the new rules [3] [89]. Conformity is demonstrated through a combination of quality management system audits and technical documentation assessment, culminating in the CE marking.
Table 1: Comparison of Core Regulatory Pathways
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | FD&C Act; 21 CFR Parts 809, 862, 864, 866 [10] | Regulation (EU) 2017/746 [3] |
| Risk Classification | 3 Classes (I, II, III) [3] | 4 Classes (A, B, C, D) [3] |
| Key Premarket Pathway | 510(k) (substantial equivalence), PMA, De Novo [10] | Conformity Assessment via Notified Body (for Classes B-D) [3] |
| "Grandfathering" Allowed | Yes (for pre-1976 devices or substantial equivalents) [3] | No (all devices must comply with IVDR) [3] [89] |
| Clinical Evidence Requirement | Required for Class III and some Class II; extent linked to risk [3] | Mandatory Performance Evaluation for all classes; depth scales with risk [3] [90] |
| Post-Market Reporting | Adverse event reporting (e.g., MDRs) [3] | Periodic Safety Update Report (PSUR) for Class C/D; Post-Market Surveillance Report (PMSR) for Class A/B [3] |
The Clinical Evidence Divide: Protocol and Data Requirements
The generation of clinical evidence represents one of the most significant resource investments. The US and EU approaches, while sharing the goal of proving safety and performance, differ in process and emphasis.
US FDA Clinical Evidence: The FDA's requirements are pathway-dependent. A 510(k) submission typically emphasizes analytical performance studies (e.g., precision, accuracy, analytical sensitivity/specificity) to demonstrate substantial equivalence to a predicate [10]. The FDA notes that while clinical information may be warranted, prospective clinical studies are rarely required for IVDs [10]. For PMA applications, robust clinical data establishing a direct link between the test result and patient health outcomes is mandatory.
EU IVDR Clinical Performance Studies: The IVDR mandates a Performance Evaluation based on three pillars: scientific validity, analytical performance, and clinical performance [3] [90]. Clinical performance studies are critical for most devices and must demonstrate that the device yields results correlating with a specific clinical condition in the target population [90]. These studies must be conducted in accordance with ISO 20916, which provides the international standard for good study practice, covering protocol design, ethics, specimen management, and reporting [90].
Table 2: Comparison of Key Clinical Evidence Requirements
| Requirement | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Core Framework | Premarket submission (510(k), PMA) specific [10]. | Performance Evaluation (PE) per Article 56 [90]. |
| Standard for Clinical Studies | Good Clinical Practice (GCP); FDA guidance documents [10]. | ISO 20916:2024 "Clinical performance studies for in vitro diagnostic medical devices" [90]. |
| Typical Study Focus for Market Entry | Analytical performance is central for 510(k); clinical outcome data for PMA [10]. | Clinical performance in real-world conditions is required for all but Class A devices [90]. |
| Specimen Source | Clinical samples, often banked or retrospective [10]. | Prospective or retrospective human specimens; strict traceability and handling per ISO 20916 [90]. |
| Post-Market Evidence | Post-approval studies may be a condition of approval. | Post-Market Performance Follow-up (PMPF) is required to update the PE continuously [3] [89]. |
The Scientist's Toolkit: Essential Reagents and Materials
Navigating dual-region compliance requires careful selection and documentation of research materials. Key reagent categories have specific regulatory definitions that impact their control and labeling.
Table 3: Key Research Reagent Solutions and Regulatory Status
| Item | Function in IVD Development | US FDA Classification | EU IVDR Status |
|---|---|---|---|
| Analyte Specific Reagent (ASR) | Antibodies, nucleic acid sequences, or ligands used to identify and quantify a specific analyte in a patient sample [10]. | Class I, II, or III medical device depending on intended use and risk [10]. | Considered an IVD device itself; classification depends on its intended purpose and risk rule application. |
| General Purpose Reagent (GPR) | Chemical reagents with broad laboratory application for specimen preparation and examination (e.g., buffers, fixatives) [10]. | Class I medical device [10]. | Typically falls under Class A (low risk) as a laboratory instrument or reagent [3]. |
| Calibrators & Controls | Materials used to establish a testing system's measurement scale and verify its ongoing accuracy and precision. | Class I or II medical devices, depending on complexity [10]. | Class A devices, unless they have a critical role for a higher-class test, which may affect their own classification. |
| Clinical Specimen Panel | Well-characterized human samples used for analytical and clinical validation studies. | Critical for study validity; must be collected under appropriate informed consent and IRB oversight [10]. | Must be managed per ISO 20916 with full traceability, documented ethical approval, and GDPR compliance [90]. |
Experimental Protocol for Dual-Region Clinical Validation
Designing studies that efficiently satisfy both FDA and IVDR requirements is paramount for resource optimization. The following protocol outlines a hybrid strategy.
Protocol: A Prospective, Multicenter Study for the Clinical Validation of a Novel Oncology IVD (Class C/PMOA)
1. Objective: To establish the clinical sensitivity, specificity, and overall agreement of the novel IVD against a validated composite clinical reference standard (histopathology + follow-up) for detecting Target Biomarker X in tissue specimens from patients with suspected Condition Y.
2. Justification for Dual-Region Applicability: This prospective design using a clinical outcome reference standard generates high-level evidence acceptable for a US PMA application. Adherence to ISO 20916 in execution ensures the data will support the Clinical Performance Study report required for the EU IVDR Performance Evaluation [90].
3. Study Design:
- Type: Prospective, blinded, multisite study.
- Sites: 10-15 clinical sites across the US and EU to ensure demographic diversity and facilitate regional regulatory acceptance.
- Sample: Consecutive eligible patients (n=###, based on power calculation) providing tissue samples during standard-of-care procedures.
- Reference Standard: A composite clinical truth adjudicated by a blinded expert committee using all available clinical data (histopathology, imaging, treatment response, 6-month follow-up).
4. Pre-Study Regulatory Engagement:
- US: Submit a Pre-Submission package to the FDA containing the draft protocol, statistical analysis plan, and key questions on acceptability of the reference standard and endpoints [10].
- EU: Initiate consultations with the targeted Notified Body to align on the suitability of the protocol, the qualifications of investigators, and the ethical review process across different Member States [90].
5. Core Experimental Workflow: The workflow for specimen and data handling is designed to meet the stringent traceability requirements of ISO 20916 and FDA GCP.
6. Key Methodology Details:
- Blinding: The testing laboratory performing the novel IVD assay and the clinical adjudication committee are blinded to each other's results.
- Specimen Management: Full chain of custody documentation from collection to disposal. Storage conditions validated and monitored.
- Data Management: Use of a 21 CFR Part 11/GDPR-compliant electronic data capture (EDC) system. Anonymization/pseudonymization procedures predefined.
- Statistical Analysis: Pre-specified analysis plan including primary endpoint calculations (sensitivity, specificity with 95% CIs) and methods for handling indeterminate/missing data.
Cost-Benefit Analysis: Strategic Resource Allocation
The divergent regulatory landscapes create distinct cost profiles and market entry timelines, directly influencing global launch strategy.
Quantitative Cost and Time Drivers
- Clinical Study Costs: EU IVDR-compliant clinical performance studies, especially prospective multicenter ones following ISO 20916, can be more expensive than studies designed solely for an FDA 510(k). However, they may be comparable or even synergistic with studies designed for a US PMA [90].
- Notified Body Fees: The EU pathway introduces substantial third-party costs from Notified Bodies for application review, audits, and certificate maintenance, which have no direct equivalent in the US FDA process [1].
- Timeline to Market: Data indicates a growing "US-First" launch strategy. The average time from application to NB certificate under IVDR can be 13-18 months, while the FDA's 510(k) process often has shorter statutory review timelines [1]. Furthermore, a 2025 survey indicated that since MDR/IVDR implementation, the choice of the EU as the first launch market fell by approximately 40% for large IVD manufacturers [1].
Table 4: Cost-Benefit Matrix for Launch Strategy
| Strategy | Estimated Upfront Cost | Time to First Revenue | Long-Term Market Access | Best For |
|---|---|---|---|---|
| US-First, EU-Follow | Lower initial outlay. EU costs deferred. | Fastest. Leverages predictable FDA pathways [1]. | Delayed EU revenue (24-36 months). Risk of NB backlog. | Start-ups, novel high-risk devices, capital-constrained firms. |
| EU-First, US-Follow | Highest initial outlay (NB fees, complex studies). | Slowest. Lengthy NB review and potential for multiple inquiry rounds [1]. | Establishes presence in large EU market first. | Devices with strong EU clinical KOL support or reimbursement advantage. |
| Parallel Submission | Very high (duplicate regulatory ops, two full submissions). | Moderate. Aims for near-simultaneous launch. | Maximizes global reach fastest. Creates complex project management. | Large, well-resourced companies with established global RA teams. |
The Strategic Pathway Decision Logic
The choice of regulatory strategy is a function of device risk, novelty, and company resources. The following diagram models the key decision points.
The comparative analysis reveals a regulatory environment where efficiency demands strategic choice, not just parallel execution. The pro-innovation stance and streamlined pathways of the US FDA often support a "US-First" launch model to accelerate time-to-revenue and generate real-world evidence [1]. In contrast, the EU's IVDR demands rigorous, upfront clinical performance data and NB interaction, creating a higher initial resource barrier [90] [1].
For researchers and developers, the optimal resource allocation strategy flows from two analyses: 1) a precise classification of the device under both systems to understand the scope of evidence required, and 2) a clear-eyed assessment of internal resources and capital. Investing in well-designed, ISO 20916-compliant clinical studies, though costly, can generate a data package that serves both regions for higher-risk devices. Ultimately, successful dual-region compliance is less about minimizing cost and more about intelligently sequencing investments to de-risk the global commercialization journey and maximize the total return across the product lifecycle. The ongoing EU initiatives to simplify regulations and the US push for AI/ML innovation will require continuous monitoring, as these policies will further shape the cost-benefit calculus in the coming years [1].
Leveraging the Pre-Submission Process (US) and Pre-Application Engagement (EU)
For developers of in vitro diagnostics (IVDs), early and strategic dialogue with regulatory bodies is a critical component of efficient product development. This guide provides a comparative analysis of the two primary formal mechanisms for this dialogue: the Pre-Submission Process with the U.S. Food and Drug Administration (FDA) and Pre-Application Engagement with European Union (EU) Notified Bodies under the In Vitro Diagnostic Regulation (IVDR). Framed within broader research on US-EU regulatory requirements, this comparison reveals that while both systems aim to de-risk the submission process, they are built upon fundamentally different regulatory philosophies and operational structures [3]. The US process is a centralized, science-led consultation with the FDA, whereas the EU process is a decentralized, conformity assessment-led dialogue with an independent Notified Body. Understanding these distinctions is essential for researchers and development professionals to navigate the complexities of global market access, optimize resource allocation, and design studies that meet the distinct evidentiary expectations of each region [91].
The Pre-Submission and Pre-Application Engagement processes are embedded within larger regulatory systems that differ in classification, centralization, and evidentiary focus.
Table 1: Foundational Comparison of US and EU IVD Regulatory Systems
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Governing Regulation | Federal Food, Drug & Cosmetic Act; 21 CFR Parts 807, 812, 814 [3]. | Regulation (EU) 2017/746 (IVDR) [3]. |
| Risk Classification | 3-tiered: Class I (lowest risk), Class II, Class III (highest risk) [3]. | 4-tiered: Class A (lowest risk), B, C, D (highest risk) [3]. |
| Reviewing Authority | Centralized (FDA's CDRH or CBER). | Decentralized (Notified Bodies designated by national authorities) [3]. |
| Primary Goal of Early Interaction | To obtain FDA feedback on specific technical, clinical, or regulatory questions to inform a future submission [92]. | To facilitate the conformity assessment process, often through a voluntary “presubmission” or “preliminary review” of documentation with a Notified Body. |
| Legal Status of Advice | Non-binding feedback from the FDA [92]. | Non-binding advice, but critical for aligning with the specific Notified Body's expectations. |
| Key Emphasis in Review | Substantial equivalence (510(k)) or safety & effectiveness (PMA/De Novo) based on analytical and clinical data [93]. | Conformity with General Safety and Performance Requirements (GSPRs), anchored by a comprehensive Performance Evaluation [3] [94]. |
Table 2: Procedural Comparison of Pre-Submission and Pre-Application Engagement
| Aspect | US FDA Pre-Submission (Q-Sub) | EU IVDR Pre-Application Engagement |
|---|---|---|
| Formal Name | Q-Submission (Pre-Sub) [55]. | Not formally standardized; often termed “pre-submission,” “presub,” or “preliminary review.” |
| Submission Format | Voluntary use of the Early Submission Requests eSTAR (PreSTAR) template (Version 2) [55]. | Determined by the individual Notified Body; no single mandated template. |
| Typical Content | Detailed questions, proposed study designs, data summaries, draft labeling [92]. | Often involves sharing draft Performance Evaluation Report, technical documentation, or quality management system procedures. |
| Formal Meeting | A written response is provided; a teleconference or face-to-face meeting can be requested [92]. | Interaction is typically via document review and correspondence; may involve structured meetings. |
| Timeline for Feedback | FDA goals are set per the Medical Device User Fee Amendments (MDUFA); typically within a set period after receipt [92]. | No legally binding timeline; subject to Notified Body capacity. Average conformity assessment timelines are reported to be around 18 months, indicating potential for delays in pre-engagement [95]. |
| Fee | Subject to FDA user fees [92]. | Subject to fees set by the individual Notified Body. |
Experimental Protocols for Generating Regulatory Evidence
The data required to support an IVD submission are generated through rigorous analytical and clinical studies. The design of these studies is often a key topic in early regulatory dialogues.
3.1 US FDA Focus: Analytical Performance for Substantial Equivalence For a 510(k) submission, the goal is to demonstrate substantial equivalence to a predicate device. The experimental protocol centers on a comparative analytical performance study [93].
- Objective: To compare the accuracy, precision, and sensitivity of the new device (test device) against a legally marketed predicate device.
- Methodology:
- Sample Selection: Procure well-characterized clinical samples (e.g., patient serum, plasma, swab eluates) spanning the assay's measuring range and covering relevant disease states and interfering conditions.
- Testing Protocol: Test all samples in parallel using the new device and the predicate device according to their respective instructions for use. Operators should be blinded to the expected results.
- Data Analysis:
- Accuracy/Bias: Calculate the correlation (e.g., Deming regression) and mean bias between the results from the two devices.
- Precision: Perform repeatability (within-run) and reproducibility (between-day, between-operator, between-lot) studies for the new device, calculating standard deviation and coefficient of variation.
- Analytical Sensitivity (LoD): Determine the lowest concentration of analyte the new device can reliably detect.
- Analytical Specificity: Evaluate interference from common substances (e.g., lipids, hemoglobin, medications) and cross-reactivity with structurally similar analytes.
- Acceptance Criteria: Predefined performance criteria should be stated, often demonstrating that the new device performs at least as well as the predicate.
3.2 EU IVDR Focus: The Comprehensive Performance Evaluation Under IVDR, the Performance Evaluation is a continuous process. The Analytical Performance Study forms its first pillar [94].
- Objective: To verify and validate the analytical performance claims listed in Annex I of the IVDR, establishing the device's technical reliability independent of clinical context [94].
- Methodology:
- Parameter-Specific Testing: Conduct discrete experiments for each applicable parameter as defined in IVDR Annex I, Section 9.1(a) [94]:
- Trueness: Comparison of measured values to a reference method or certified reference material.
- Precision: Repeatability and reproducibility studies as per CLSI guidelines.
- Analytical Sensitivity: Determination of Limit of Blank (LoB), Limit of Detection (LoD), and Limit of Quantitation (LoQ) for quantitative assays.
- Measuring Range & Linearity: Demonstration that the device provides valid results across the claimed range.
- Analytical Specificity: Assessment of interference and cross-reactivity.
- Sample Considerations: Use of clinical samples is expected, but the study is designed to characterize the device's technical performance, not its clinical diagnostic accuracy.
- Statistical Justification: The sample size for each experiment must be justified statistically to ensure the reliability of the estimates.
- Parameter-Specific Testing: Conduct discrete experiments for each applicable parameter as defined in IVDR Annex I, Section 9.1(a) [94]:
- Integration: Results are compiled into an Analytical Performance Report (APR), which, alongside the Clinical Performance Report and evidence of Scientific Validity, forms the Performance Evaluation Report (PER) [94].
Diagram: US FDA Pre-Submission (Q-Sub) Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for IVD Performance Studies
| Item | Primary Function | Considerations for US vs. EU |
|---|---|---|
| Certified Reference Materials (CRMs) | To establish trueness and calibrate assays by providing an unbroken metrological traceability chain to a higher-order standard. | Critical for EU IVDR to demonstrate metrological traceability (Annex I GSPR 9.1(b)) [94]. Used in US for calibration but may have less emphasis on formal traceability in 510(k). |
| Well-Characterized Clinical Samples | To assess analytical and clinical performance across relevant biological matrices, disease states, and concentrations. | Core for both regions. For US 510(k), samples are used for direct comparison to a predicate. For EU, they underpin both Analytical and Clinical Performance Reports [94] [93]. |
| Interferent Stocks | To evaluate analytical specificity by spiking samples with potential interfering substances (e.g., bilirubin, lipids, common drugs). | Required for both. Protocols should reflect endogenous and exogenous substances relevant to the sample type and intended use. |
| Panel for Cross-Reactivity | To assess specificity against genetically or structurally related analytes (e.g., different virus strains, homologous proteins). | Required for both. The breadth of the panel should be justified based on the risk of cross-reaction and intended use claims. |
| Stability Study Materials | To establish shelf-life, in-use stability, and transport conditions under defined temperatures and humidity. | Required for both. IVDR explicitly references stability standards (e.g., ISO 23640) and requires real-time data for Class C/D devices where possible [94]. |
Diagram: EU IVDR Pre-Application Engagement Process
The comparative analysis underscores a fundamental dichotomy: the US Pre-Submission is a predictable, centralized interaction with the regulator on scientific and regulatory strategy, while the EU Pre-Application Engagement is a variable, critical negotiation with a commercial conformity assessment body on the totality of technical documentation. For researchers, this has direct implications. A development program targeting both markets must be designed to satisfy the FDA's focus on controlled comparative studies and the IVDR's expansive requirements for stand-alone, scientifically valid performance evidence across three pillars [3] [94]. Furthermore, the EU's ongoing challenges—such as Notified Body capacity constraints, lack of binding review timelines, and evolving guidance—introduce a significant element of timing uncertainty that is less pronounced in the more structured US system [95]. Consequently, a strategic global regulatory plan should not only sequence scientific activities but also account for the longer and less predictable EU engagement and assessment pathways. Early and parallel investment in building IVDR-compliant Performance Evaluation documentation is essential to avoid it becoming a critical path item for market access in Europe.
Synthesizing a Global Regulatory Strategy for Efficient Market Entry
The global in vitro diagnostics (IVD) market is defined by a profound and widening regulatory divergence between its two largest markets: the United States and the European Union. This divergence presents a critical strategic challenge for researchers and product developers. Where the U.S. maintains a pro-innovation stance characterized by flexible pathways for novel technologies like AI, the EU enforces a precautionary, complex system under the In Vitro Diagnostic Regulation (IVDR) and Medical Device Regulation (MDR) [1]. This regulatory divide directly influences time-to-market, development costs, and global launch sequencing. Within this context, a strategic comparison is not merely academic but a practical necessity for efficient global market entry. This guide provides an evidence-based comparison of U.S. and EU IVD regulatory requirements, framing the analysis within the broader thesis that understanding this divergence is foundational to building a viable global regulatory strategy.
Comparative Analysis: US FDA vs. EU IVDR Frameworks
Foundational Philosophies and Market Consequences
The core philosophies of the two regulatory systems have created tangible market outcomes. The U.S. FDA operates as an active partner in technological progress, emphasizing a predictable and collaborative review process [1]. In contrast, the EU's IVDR prioritizes a high level of patient safety through a stringent, ex-ante conformity assessment model overseen by Notified Bodies [3]. This philosophical clash has significant commercial implications: the U.S. now holds approximately 46.4% of the global MedTech market, while Europe's share is 26.4% [1]. Surveys indicate that since the implementation of MDR/IVDR, the choice of the EU as a first launch market has dropped by approximately 40% for large IVD manufacturers [1].
Classification Systems and Approval Pathways
A primary difference lies in risk classification and the corresponding routes to market.
Table 1: Comparison of US and EU IVD Risk Classification and Pathways
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Regulatory Basis | Regulated as medical devices under FD&C Act, 21 CFR [3] | Regulated under distinct IVD Regulation (EU) 2017/746 [3] [11] |
| Risk Classes | Class I (lowest risk), Class II, Class III (highest risk) [3] | Class A (lowest risk), Class B, Class C, Class D (highest risk) [3] |
| Key Market Pathway | Premarket Notification [510(k)] demonstrating substantial equivalence to a predicate; Premarket Approval (PMA) for Class III [96]. | Conformity assessment requiring Notified Body involvement for most classes (B, C, D); Self-declaration for some Class A devices [3] [96]. |
| Equivalence Route | Foundation of the 510(k) pathway; well-established [96]. | Possible but strict criteria for "exact equivalence"; considered difficult under IVDR [1] [96]. |
| Validity Period | Approval is valid indefinitely unless significant changes are made [96]. | Notified Body certificates issued for a maximum of 5 years [96]. |
Core Regulatory Requirements and Timelines
The substantive requirements for performance evaluation, quality management, and post-market vigilance also differ significantly.
Table 2: Comparison of Key Regulatory Requirements and Processes
| Aspect | United States (FDA) | European Union (IVDR) |
|---|---|---|
| Clinical Evidence | Required for Class III and some Class II devices; scope tied to submission pathway [3]. | Required for all IVDs via a Performance Evaluation (Analytical, Clinical, Scientific Validity) [3]. |
| Quality System | Quality System Regulation (QSR, 21 CFR Part 820). Transitioning to QMSR (harmonized with ISO 13485) by Feb 2026 [23] [50]. | QMS required per Article 10(8) IVDR, must be certified to ISO 13485 for NB-involved assessments [96]. |
| Post-Market Surveillance | Mandatory reporting of adverse events (MDR) [3]. | Structured PMS system; Periodic Safety Update Reports (PSURs) for Class C/D, PMS Reports for Class A/B [3]. |
| Unique Device ID (UDI) | Required, submitted to FDA's GUDID database [3]. | Required, format differs from US; data submitted to EUDAMED [3]. |
| Typical Procedure Duration | 510(k): ~3-9 months; De Novo: ~1-1.5 years; PMA: ~1.5-2 years [96]. | Notified Body process: >12-18 months commonly cited due to NB shortages and application complexity [1] [96]. |
| Typical Authority Costs | 510(k): ~$20,000; PMA: ~$440,000 (lower for small businesses) [96]. | Notified Body fees: €30,000 to €100,000+, highly variable [96]. |
Strategic Implications for Product Development and Launch
The Solidified "US-First" Launch Model
The comparative data supports a dominant "US-First" launch strategy. The U.S. pathway offers faster, more predictable access for many devices, enabling early revenue generation and real-world evidence collection [1]. This evidence can later be leveraged in a more challenging EU submission. The U.S. pro-innovation policy is exemplified by the FDA's Predetermined Change Control Plan (PCCP) for AI/ML devices, allowing pre-approved iterative updates without new submissions—a stark contrast to the EU's rigid system [1].
Navigating the EU's "Chronic Challenge"
Despite its challenges, the EU remains an essential market. Strategic entry requires understanding its evolving timeline. Transition deadlines have been extended to avert a crisis: Class D IVDs must comply by December 2027, with Classes C, B, and A (sterile) following through 2028-2029 [1] [28]. However, systemic constraints persist. As of early 2025, only about 12,177 MDR certificates had been issued from over 28,489 applications, with processing taking 13-18 months for 60% of cases [1]. Proactive preparation and engagement with a Notified Body are critical.
The AI and Cybersecurity Compliance Front
For software and AI-driven IVDs, the regulatory chasm is most pronounced.
- AI Governance: The EU subjects AI medical devices to a mandatory, high-risk classification under the AI Act, integrated with IVDR requirements [1]. The U.S. promotes a voluntary, flexible framework via the NIST AI Risk Management Framework (RMF), though FDA expects robust algorithm validation [1] [23].
- Cybersecurity: The FDA mandates a Secure Product Development Framework (SPDF) and a Software Bill of Materials (SBOM) for premarket submissions [1]. The EU's requirements are multi-layered, stemming from IVDR's safety requirements, the NIS 2 Directive, the AI Act, and the Radio Equipment Directive [1].
Publish Comparison Guides: Experimental Data on Regulatory Performance
Comparative Study: Time-to-Market for a Novel Molecular IVD
This simulated study compares the regulatory journey of a novel Class C (EU) / Class II (US) molecular diagnostic from submission to authorization.
Table 3: Simulated Time-to-Market Comparison for a Class C/II IVD
| Phase | U.S. FDA (510(k) Route) | EU IVDR (Notified Body Route) | Supporting Data / Rationale |
|---|---|---|---|
| Pre-Submission | 3 months (Pre-Sub meeting, final packaging) | 6+ months (NB selection, gap analysis, QMS audit prep) | EU requires full QMS certification audit if not already obtained [96]. |
| Formal Review | 90-150 calendar days (FDA review clock) | 12-18 months (NB review, sample testing, QMS audit) | Published averages for NB review times [1] [96]. |
| Interactive Review | 30-60 days (Responding to FDA Additional Information requests) | 3-6 months (Addressing NB questions, compiling additional clinical data) | EU's clinical evidence requirements under IVDR are more extensive [3]. |
| Total Estimated Time | 6-9 months | 21-30 months | The U.S. pathway is estimated to be ~15-21 months faster. |
Methodology: This comparison is constructed from published industry averages for regulatory procedure durations [1] [96], accounting for the prerequisite of a certified QMS under IVDR and the typical interactive review phases in both jurisdictions. It assumes a well-prepared submission with a valid predicate for the U.S. and no requirement for a new clinical performance study in the EU.
Performance Evaluation: Stringency of Clinical Evidence Requirements
An analysis of the depth and breadth of clinical evidence required reveals a key strategic differentiator.
Table 4: Comparison of Clinical Evidence Requirements
| Parameter | U.S. FDA (for a 510(k) with Predicate) | EU IVDR (for a Class C Device) |
|---|---|---|
| Basis of Claim | Substantial equivalence to a predicate device. | Independent demonstration of safety and performance per Annex XIII. |
| Data Acceptability | Existing literature, retrospective studies, and predicate data often suffice. | Heavy reliance on prospective clinical performance studies is common, especially for novel markers [3]. |
| Ongoing Requirement | Post-market surveillance and potential post-approval studies for certain devices. | Continuous process: Performance Evaluation Report is updated via Post-Market Performance Follow-up (PMPF) [3]. |
| Regulatory Flexibility | Accepted use of real-world data and bridging studies when scientifically justified, especially for rare biomarkers [23]. | Less flexibility; common specifications for Class D devices and expectations for clinical studies create a higher barrier [11]. |
Experimental Protocol for Generating EU IVDR Clinical Performance Data:
- Study Design: A prospective, multi-center clinical performance study.
- Objective: To establish clinical sensitivity and specificity against a pre-defined clinical truth standard (e.g., PCR, sequencing).
- Sample Size Calculation: Powered to achieve confidence intervals for sensitivity/specificity with a width no greater than 10%.
- Site Selection: Minimum of 3-5 independent clinical sites within the EEA to ensure representativeness.
- Analysis: Performance metrics calculated with 95% confidence intervals. Results integrated into the Performance Evaluation Report, which links analytical data to scientific validity and clinical performance.
Strategic Pathway Visualization
Strategic Pathways for US and EU IVD Market Entry
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 5: Key Regulatory and Compliance Resources
| Tool / Resource | Primary Function | Relevance to Strategy |
|---|---|---|
| ISO 13485:2016 QMS | Provides the framework for a quality management system harmonized with regulatory requirements [28] [96]. | Fundamental for EU compliance and increasingly for US (QMSR). Early certification accelerates both US and EU pathways. |
| EUDAMED (European Database) | The EU's central database for device registration, UDI, NB certificates, clinical investigations, vigilance, and market surveillance [3] [11]. | Mandatory for EU market access. Engaging with its "playground" environment for testing is advised ahead of mandatory use [23]. |
| FDA Guidance Documents & Recognized Consensus Standards | Clarify FDA's interpretation of regulations and list recognized standards (e.g., CLSI, ISO) for demonstrating compliance [96]. | Critical for efficient US submission preparation. Following recognized standards streamlines review. |
| IVDR Annex I (GSPRs) Checklist | A detailed checklist derived from the General Safety and Performance Requirements of the IVDR. | Essential for systematically building the EU Technical Documentation and ensuring all mandatory requirements are addressed. |
| Clinical Performance Study Protocol Template | A standardized template for designing studies that meet IVDR Annex XIII and XIV requirements. | Ensures generated clinical evidence will be acceptable to Notified Bodies, reducing the risk of costly study repetition. |
| Regulatory Intelligence Platform | A subscription service (e.g., from commercial providers) tracking real-time changes in global regulations, guidance, and standards. | Enables proactive strategy adjustments in a fast-evolving landscape, particularly for AI and cybersecurity [1] [23]. |
Synthesis and Strategic Framework Visualization
Decision Framework for Global IVD Market Entry Strategy
The synthesized strategy is clear. For most novel IVDs, particularly those involving software and AI, the evidence supports a US-First launch model to capitalize on faster market access, a collaborative regulator, and pathways for iterative improvement [1]. The EU IVDR demands a separate, resource-intensive, and longer-term compliance project focused on Notified Body engagement, comprehensive performance evaluation, and lifecycle vigilance [3] [96]. Success hinges on understanding this dichotomy not as a barrier but as a map—one that, when read correctly, allows researchers and developers to sequence their global entry efficiently, using data from one jurisdiction to strategically support applications in the other.
Conclusion
The US and EU IVD regulatory landscapes represent a significant strategic divergence, with the US fostering a pro-innovation environment through mechanisms like the Predetermined Change Control Plan for AI, while the EU prioritizes a rigorous, precautionary framework under IVDR. This has solidified a 'US-First' launch model for many innovators, allowing for early revenue and real-world evidence generation before tackling the more complex EU pathway. Key takeaways for researchers and developers include the critical need for early and distinct strategic planning for each market, a deep understanding of the expanded clinical evidence and post-market surveillance requirements under IVDR, and proactive management of new rules for LDTs and AI-driven diagnostics. Future success will hinge on agile regulatory strategies that can adapt to ongoing reforms, such as the EU's efforts to simplify MDR/IVDR, and the growing convergence of digital health, AI, and diagnostics, demanding robust lifecycle management for global market access.
References
- 1. The 2025 MedTech Regulatory Divide: A Strategic Analysis ... [https://htec.com/insights/a-strategic-analysis-of-the-2025-us-eu-medtech-regulatory-divide/]
- 2. FDA Continues to Strengthen the 510(k) Program [https://www.fda.gov/medical-devices/510k-clearances/fda-continues-take-steps-strengthen-premarket-notification-510k-program-program-updates]
- 3. IVDs – A Comparison of Requirements between the US and EU [https://blog.pqegroup.com/medical-device/ivds-a-comparison-of-requirements-between-the-us-and-eu]
- 4. Premarket Notification 510(k) [https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k]
- 5. Comparing FDA's 510(k) and EU's MDR frameworks for ... [https://www.linkedin.com/posts/steveleeivd_healthtech-regulation-510k-activity-7325068924591513600-iahI]
- 6. EU MDR vs FDA: 2025 US-EU Medical Device Regulatory ... [https://www.complizen.ai/post/eu-mdr-vs-fda-complete-regulatory-comparison-guide-2025]
- 7. Clinical Study Management for FDA and EU Submissions [https://www.mdcassoc.com/clinical-study-management-for-fda-and-eu-submissions/]
- 8. Safety and Performance Based Pathway [https://www.fda.gov/medical-devices/premarket-notification-510k/safety-and-performance-based-pathway]
- 9. European Commission launches 'Call for Evidence' on the ... [https://www.medtecheurope.org/2025/10/02/european-commission-launches-call-for-evidence-on-the-future-of-the-medical-devices-regulation-and-in-vitro-diagnostics-regulation/]
- 10. Overview of IVD Regulation [https://www.fda.gov/medical-devices/ivd-regulatory-assistance/overview-ivd-regulation]
- 11. New Regulations - Public Health - European Commission [https://health.ec.europa.eu/medical-devices-sector/new-regulations_en]
- 12. US FDA vs EU MDR: Device Classification Differences [https://cmcmedicaldevices.com/us-fda-vs-eu-mdr-key-differences-in-medical-device-classification/]
- 13. Regulatory landscape of accelerated approval pathways for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12119601/]
- 14. Comparison of the international regulations for medical ... [https://www.sciencedirect.com/science/article/pii/S0020138323005946]
- 15. International Medical Device Regulators Forum (IMDRF) [https://www.fda.gov/medical-devices/cdrh-international-affairs/international-medical-device-regulators-forum-imdrf]
- 16. Notified bodies for medical devices - Public Health [https://health.ec.europa.eu/medical-devices-topics-interest/notified-bodies-medical-devices_en]
- 17. EU Notified Bodies Survey 2025 on MDR and IVDR ... [https://www.pureglobal.com/news/eu-notified-bodies-survey-2025-on-mdr-and-ivdr-certifications-and-applications]
- 18. IVDR Deadline: Class D IVDs Need Notified Body ... [https://www.obelis.net/news/september-2025-eu-deadline-for-ivds-signed-agreement-for-class-d-legacy-devices/]
- 19. Top 5 Upcoming FDA and EU Regulations - MEDIcept [https://www.medicept.com/top-5-upcoming-fda-and-eu-regulations-what-to-know-for-2025/]
- 20. IVDR Classification for In Vitro Medical Devices: An Explainer [https://www.greenlight.guru/blog/ivdr-classification]
- 21. How to Classify Your Medical Device Under the EU MDR ... [https://www.arenasolutions.com/resources/articles/classify-medical-device-eu-mdr-ivdr/]
- 22. CE Marking for Medical Devices: Definition, Requirements, ... [https://simplerqms.com/ce-marking-for-medical-devices/]
- 23. IVD Regulatory Update 2025 from AMDM Meeting [https://beaufortcro.com/blog/ivd-regulatory-update-2025/]
- 24. US FDA vs EU Medical Device Classification Guide [https://www.emergobyul.com/news/us-fda-and-eu-risk-classification-medical-devices]
- 25. EU CE Marking Approval for IVDs [https://www.pureglobal.com/markets/european-union/eu-ivdr-consulting/ce-marking-approval-for-ivds]
- 26. EU Releases Guidance on IVD Performance Studies, Finally [https://www.emergobyul.com/news/eu-releases-guidance-ivd-performance-studies-finally]
- 27. Mastering IVDR in 2025: Challenges and Strategies Across ... [https://www.ul.com/resources/mastering-ivdr-2025-challenges-and-strategies-across-full-compliance-spectrum]
- 28. Navigating the IVD Regulatory Landscape in 2025 - Bioexcel [https://bioexcelife.com/navigating-the-ivd-regulatory-landscape-in-2025/]
- 29. Trends and Prospects Shaping Europe's US$34.47 Billion ... [https://finance.yahoo.com/news/trends-prospects-shaping-europes-us-102800009.html]
- 30. Guide to Premarket Approval (PMA) vs. De Novo [https://namsa.com/resources/blog/approval-pma-vs-de-novo/]
- 31. 510(k) vs De Novo: Key Differences in FDA Approval ... [https://www.freyrsolutions.com/blog/510k-vs-de-novo-key-differences-in-fda-approval-pathways-for-medical-devices]
- 32. Beyond the 510(k): The regulation of novel moderate-risk ... [https://www.nature.com/articles/s41746-024-01021-y]
- 33. FDA vs IVDR Companion Diagnostics [https://mdxcro.com/fda-vs-eu-ivdr-companion-diagnostics-regulation/]
- 34. MDCG 2025-5 Clarifies EU Rules for IVD Performance ... [https://www.pureglobal.com/blog-posts/mdcg-2025-5-clarifies-eu-rules-for-ivd-performance-studies-under-ivdr]
- 35. A Comparison of IVDR to FDA IVD Regulatory Submission ... [https://www.rqmplus.com/blog/a-comparison-of-ivdr-to-fda-ivd-regulatory-submission-requirements/]
- 36. 2. Notified Bodies and Conformity assessment [https://learning.eupati.eu/mod/page/view.php?id=936]
- 37. IVDR Classification Guidance [https://www.distillersr.com/resources/meddev-literature-review/ivdr-classification-guidance]
- 38. EU IVDR CE Marking Regulatory Process Chart [https://www.emergobyul.com/resources/european-vitro-diagnostic-devices-regulation-ivdr-ce-marking-regulatory-process]
- 39. How Do You Comply with EU MDR/IVDR vs. U.S. FDA ... [https://avendium.com/2025/10/31/how-do-you-comply-with-eu-mdr-ivdr-vs-u-s-fda-regulations/]
- 40. Medical Device Technical File Requirements [https://www.cognidox.com/blog/medical-device-technical-file-requirements-what-you-need-to-know]
- 41. EU MDR vs. IVDR: What are the differences? [https://advisera.com/13485academy/blog/2021/04/06/ivdr-vs-mdr-comparison/]
- 42. In Vitro Diagnostics Market to Hit USD 197.06 Billion by ... [https://www.biospace.com/press-releases/in-vitro-diagnostics-market-to-hit-usd-197-06-billion-by-2032-driven-by-a-surge-in-chronic-and-infectious-disease-burden-coherent-market-insights]
- 43. Performance Evaluation under the IVDR:A Practical Overview [https://decomplix.com/performance-evaluation-ivdr-practical-overview/]
- 44. The EU IVDR: An Overview of Regulatory Requirements in 2017/746 [https://www.orielstat.com/blog/eu-ivdr-changes-2017-746/]
- 45. In Vitro Diagnostics - North America | Market Forecast [https://www.statista.com/outlook/hmo/medical-technology/in-vitro-diagnostics/north-america?srsltid=AfmBOooX-gQVBRv-r8bKLnWrcoAM52okxwZksvJ1-PzCAnsC42vBzP-g]
- 46. Regulation (EU) 2017/746 (EU IVDR) [https://euivdr.com/]
- 47. Quality Management System Regulation: Final Rule [https://www.fda.gov/medical-devices/quality-system-qs-regulationmedical-device-current-good-manufacturing-practices-cgmp/quality-management-system-regulation-final-rule-amending-quality-system-regulation-frequently-asked]
- 48. 2025 List: Harmonized Standards/Common Specifications [https://casusconsulting.com/list-mdr-ivdr-harmonized-standards-common-specifications/]
- 49. Adapting your QMS to IVDR requirements until May 2025 [https://www.pureglobal.com/blog-posts/adapting-your-qms-to-ivdr-requirements-until-may-2025-what-manufacturers-need-to-know]
- 50. FDA medical device inspections in 2025: What we're ... [https://www.hoganlovells.com/en/publications/fda-medical-device-inspections-in-2025]
- 51. Cultural shift of ISO 13485:2016 implementation for the FDA [https://www.iconplc.com/insights/blog/2025/05/21/cultural-shift-iso-13485-2016-implementation-fda]
- 52. Why do I still need to look at my QMS under the IVDR if it is ... [https://www.qbdgroup.com/en/blog/why-do-i-still-need-to-look-at-my-qms-under-the-ivdr-if-it-is-iso-13485-certified]
- 53. Stick or Twist: ISO 13485 Compliance Changes in 2025 [https://www.rqmplus.com/blog/stick-or-twist-2025-is-a-big-year-for-iso-13485/]
- 54. FDA Finalizes Guidance on Emergency Use of IVDs During ... [https://www.pureglobal.com/news/fda-finalizes-guidance-on-emergency-use-of-ivds-during-public-health-crises-2025]
- 55. eSTAR Program [https://www.fda.gov/medical-devices/how-study-and-market-your-device/estar-program]
- 56. FDA vs EU: Regulations compared [https://www.we4cr.com/smart-clinical-trials-fda-vs-eu-2025/]
- 57. IVDD to IVDR: A Sponsor's Guide to Europe's Diagnostic ... [https://www.lindushealth.com/blog/ivdd-to-ivdr-a-sponsors-guide-to-europes-diagnostic-revolution]
- 58. EU IVDR: Ensure compliance with technical documentation ... [https://medfilesgroup.com/eu-ivdr-technical-documentation-notified-body-transition-periods/]
- 59. DNV recognized as a Notified Body for Certification under In ... [https://www.dnv.com/news/2025/ivdr-press-release/]
- 60. IVDR 2025 Deadline: Key Requirements for Legacy IVDs ... [https://www.obelis.net/news/ivdr-2025-legacy-deadline/]
- 61. (EU) 2024/1860 – IVDR 26 September 2025 deadline ... [https://www.bsigroup.com/en-US/insights-and-media/media-center/press-releases/2025/july/eu-20241860--ivdr-26-september-2025-deadline-approaching--impact-on-legacy-devices-not-transitioning-to-the-ivdr/]
- 62. IVDR: Navigating supply chain challenges with confidence [https://www.dnv.com/supplychain/insights/ivdr-navigating-supply-chain-challenges-with-confidence/]
- 63. Laboratory Developed Tests [https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests]
- 64. FDA Reverses Final Rule on LDTs: A Win for Labs, a Shift ... [https://www.swlaw.com/publication/fda-reverses-final-rule-on-ldts-a-win-for-labs-a-shift-in-regulatory-strategy/]
- 65. The LDT Final Rule Bites the Dust [https://www.ebglaw.com/insights/publications/the-ldt-final-rule-bites-the-dust-examining-the-repercussions-of-the-federal-courts-vacatur-and-what-the-future-may-hold]
- 66. How to Comply with Stage 1 of FDA's LDT Final Rule [https://www.thefdagroup.com/blog/how-to-comply-with-stage-1-of-fda-ldt-final-rule]
- 67. Understanding the Evolving Landscape of IVD Device ... [https://www.intertek.com/blog/2025/09-04-understanding-ivd-device-compliance-and-safety/]
- 68. Bridging the IVD Regulatory Gap in Clinical Trials [https://www.qbdgroup.com/en/blog/ivd-clinical-trials-integration]
- 69. What's the difference between selling medical devices in ... [https://medenvoyglobal.com/blog/whats-the-difference-between-selling-medical-devices-in-the-us-and-europe/]
- 70. Comparison of MDR/IVDR and EU AI Act Requirements [https://www.daliag.com/post/comparison-of-mdr-ivdr-and-eu-ai-act-requirements]
- 71. Updated Post-Market Surveillance Requirements Now in ... [https://compliancenavigator.bsigroup.com/en/medicaldeviceblog/great-britains-updated-post-market-surveillance-requirements-now-in-force-what-manufacturers-need-to-know/]
- 72. Medical Device Post-Market Surveillance: Complete FDA ... [https://www.complizen.ai/post/medical-device-post-market-surveillance-fda]
- 73. Global trends in post-market surveillance of high-risk ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12624750/]
- 74. Market surveillance and vigilance - Public Health [https://health.ec.europa.eu/medical-devices-sector/directives/market-surveillance-and-vigilance_en]
- 75. 2025 Q3: Market Data Transparency and Regulatory ... [https://www.emergobyul.com/news/2025-q3-commitments-market-data-transparency-and-regulatory-reliance]
- 76. New EU Guidance on AI Act, MDR, and IVDR Compliance [https://advenamedical.com/new-eu-guidance-on-ai-act-mdr-and-ivdr-compliance/]
- 77. The differences between the US FDA's device approval ... [https://mender.io/blog/the-differences-between-fda-and-mdr-regulations]
- 78. Decoding MDCG 2025-6: Interplay between the MDR/IVDR ... [https://www.emergobyul.com/news/decoding-mdcg-2025-6-interplay-between-mdrivdr-and-aia]
- 79. Navigating the EU AI Act: implications for regulated digital ... [https://www.nature.com/articles/s41746-024-01232-3]
- 80. What the AI Act means for medical device and IVD ... [https://blog.johner-institute.com/iec-62304-medical-software/ai-act-eu-ai-regulation/]
- 81. AI Medical Device Software under EU MDR & IVDR [https://decomplix.com/ai-medical-device-software-eu-mdr-ivdr/]
- 82. 21 CFR Part 801 -- Labeling [https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-801]
- 83. UDI Basics [https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/udi-basics]
- 84. UDI for Medical Devices: Codes & Examples [Ultimate Guide] [https://www.greenlight.guru/blog/udi-medical-devices]
- 85. Unique Device Identifier - UDI - Public Health [https://health.ec.europa.eu/medical-devices-topics-interest/unique-device-identifier-udi_en]
- 86. UDI Differences – US and EU [https://www.tracekey.com/en/udi-differences-eu-us/]
- 87. Unique Device Identification System (UDI System) [https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/unique-device-identification-system-udi-system]
- 88. UDI & Class I Medical Devices: The obligation is now in ... [https://www.certeafiles.io/en/blog/udi-classe-un]
- 89. IVD Manufacturers & EU IVD Regulation Changes (May ... [https://www.precisionformedicine.com/blog/ivd-manufacturers-eu-ivd-regulation-changes-may-2022/]
- 90. Running Clinical Studies Under IVDR: What You Need to ... [https://mdxcro.com/running-clinical-studies-under-ivdr-what-you-need-to-know/]
- 91. Comparing US FDA vs EU MDR Medical Device Software ... [https://namsa.com/resources/blog/comparing-us-fda-vs-eu-mdr-medical-device-software-requirements/]
- 92. De Novo Classification Request [https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/de-novo-classification-request]
- 93. In Vitro Diagnostic (IVD) Devices Complete Guide ... [https://www.greenlight.guru/blog/ivd-devices]
- 94. Performance evaluation according to IVDR: analytical ... [https://www.seleon.com/en/performance-evaluation-according-to-ivdr-analytical-performance/]
- 95. EU Medical Devices Legislation: What You Need To Know ... [https://www.arnoldporter.com/en/perspectives/advisories/2025/03/eu-medical-devices-legislation-what-you-need-to-know]
- 96. Medical device approval EU and USA [https://blog.johner-institute.com/regulatory-affairs/medical-device-approval-approval-procedures-in-the-eu-and-the-usa/]